
Uncategorized
Вроджені катаракти
(Congenital Cataracts)
С.О. Поліщук
Лікар-генетик Волинського обласного
дитячого територіального медичного об’єднання
Основні діагностичні критерії:
Катаракти – це помутніння кришталика, які супроводжуються зниженням зору від незначного зниження до світловідчуття. Деякі дослідники вважають, що непрогресуючі ізольовані мікропомутніння кришталика, які не знижують гостроти зору не повинні називатися катарактами крім відомих асоціацій з метаболічними чи спадковими хворобами.
Клінічна картина:
Клінічна картина вроджених катаракт дуже варіабельна в залежності від інтенсивності, форми і локалізації помутніння кришталика, а також наявності чи відсутності інших дефектів ока. Вроджені катаракти можуть бути класифіковані відповідно до їх морфології:
- передня полярна;
- передня кортикальна;
- ядерна;
- ламелярна;
- повна (дифузна);
- секторальна;
- задня кортикальна;
- задній лентікоконус (лентікоглобус).
Для діагностики катаракт найчастіше використовується біомікроскопічне обстеження з допомогою щілинної лампи. Метод дозволяє діагностувати помутніння кришталика, визначити їх розміри, інтенсивність і локалізацію, а також обстежити передню і задню капсули. Використовують також обстеження з використанням прохідного і бокового освітленням та офтальмоскопію.
Полярна катаракта (передня і задня) являє собою округле, щільне, білувато-сіре помутніння капсули і прилягаючих шарів кришталика.
В прохідному світлі, на фоні рожево-червоного рефлексу з очного дна видно округле, темне, як правило, невелике помутніння. Полярні катаракти або не впливають на гостроту зору, або ненабагато знижують її.
Повна (дифузна) катаракта характеризується тотальним помутнінням кришталика, форма і розміри якого зберігаються. Рефлекс з очного дна відсутній як при вузькій так і при розширеній зіниці. Зір знижений до світловідчуття.
Ламелярна катаракта – це часткове помутніння кришталика у вигляді окремих шарів, розміщених між ембріональним ядром і кортикальними шарами. Помутніння має форму диска діаметром 3 – 9 мм з чіткими рівними краями. По екватору диск може мати помутніння перинуклеарної зони (так звані “наїздники”), які мають форму петлі або гачка. Гострота зору в межах від декількох сотих до декількох десятих, досягаючи інколи 0,3 – 0,5, повільно падає і призводить до інвалідності по зору.
Ядерна катаракта характеризується помутнінням ембріонального ядра кришталика. При боковому освітленні катаракта має вигляд невеликого сірого диска в центрі кришталика, в прохідному світлі на фоні рожево-червоного рефлексу з очного дна видно центральний темний диск. Гострота зору значно знижена.
Інші форми часткового помутніння кришталика, особливо в ділянці осі кришталика, уражають декілька шарів, не прогресують і на гостроту зору не впливають.
Ускладнення:
Амбліопія, ністагм, страбізм, відсутність або значне зниження гостроти зору.
Поєднання з іншими вродженими вадами:
Більшість вроджених катаракт поєднані з іншими аномаліями ока. Серед них: глаукома, відшарування сітківки, мікрофтальм, колобоми судинного тракту і зорового нерву, зміщення зіниці, ністагм, страбізм, мікрокорнеа, помутніння скловидного тіла, атрофія зорового нерва, дизгенез райдужки і рогівки, персистуюче гіперпластичне скловидне тіло.
Катаракти також діагностують при численних метаболічних хворобах: цукровому діабеті, гепатолентикулярній дегенерації, окуло-церебро-ренальному синдромі, хворобі Фабрі, галактоземії, гомоцистинурії.
Катаракти також зустрічаються при хромосомних абераціях (найчастіше – хромосоми 21-ї трисомія, хромосоми 13-ї трисомія, хромосоми 18-ї трисомія).
Дерматологічні хвороби, такі, як іхтіоз, синдром нетримання пігменту, пойкілодермія і актодермальна дисплазія, також можуть бути поєднані з катарактами.
Етіологія:
Катаракти етіологічно різноманітні і описані при багатьох менделюючих, тератогенних і хромосомних синдромах. Більшість з них мають аутосомно-домінантний тип успадкування, рідше, аутосомно-рецесивний та Х-зчеплений тип. Катаракти, які є складовою частиною множинних мальформаційних синдромів, успадковуються відповідно таким же чином, як і синдроми. З катарактою у плода також асоціюються вживання матір’ю алкоголю та наркотиків, інфекції, такі як краснуха під час внутріутробного розвитку. В цих спорадичних випадках можна побачити всі форми катаракт, але найчастіше – повну, мембранозну, дископодібну.
Патогенез:
Катаракти – це гетерогенна група захворювань, з яких приблизно 25% спадково обумовлені. Про патогенез вроджених катаракт відомо мало. Окремі помутніння можуть утворитися з недостатньо сформованих волокон кришталика і переродитися в точкові катаракти.
Виявлено, що деякі помутніння кришталика можуть бути утворені цистеїном, тирозином і кальцієм. Дископодібні катаракти можуть виникнути від втручань в розвиток на 5-му місяці вагітності.
Повні катаракти можуть мати відношення до персистуючої артерії скловидного тіла, порушення процесу відшарування кришталикового пухирця від зовнішньої ектодерми або внутріматкового запалення. Метаболічні фактори включають порушення обміну кальцію та галактоземію.
Утворення передньої полярної катаракти пов’язують з наявністю залишків зіничної мембрани.
Співвідношення статей: Ч1 : Ж1.
Популяційна частота:
Невеликі помутніння кришталика можна знайти приблизно у 50% населення. Але клінічно значущі помутніння кришталика зустрічається з частотою 1 на 250 новонароджених і генетична етіологія може бути визначена приблизно в 25% цих випадків (за даними США). Огляди школярів в школах для дітей з поганим зором показали, що 18-25% з них мають або мали катаракти.
Повторний ризик для сибсів пробанда:
При катаракті оцінюється як для аутосомно-домінантного типу, рідше як для аутосомно-рецесивного і Х-зчепленого, враховуючи неоднорідність в генетичному і патогенетичному відношенні.
Профілактика:
Рекомендоване медико-генетичне консультування з детальним аналізом родоводу, профілактика катаракт при галактоземії (дефіциті галактозо-епімерази, галактокінази), уникнення негативного впливу запідозрених тератогенів, проведення оцінки діагностичних тестів.
Лікування:
Екстракція катаракти по показах, корекція афакії і амбліопії.
Прогноз:
Сприятливий при ізольованому і нещільному помутнінні кришталика.
Визначення носійства:
В більшості випадків невідоме. Ген вродженої повної катаракти локалізований на Х хромосомі, ген зонулярної порошкоподібної катаракти (Х-зчепленої) – на 1q21 – 1q25.
Синдроми, при яких спостерігається катаракта
| Синдром | Інші симптоми | Етіологія |
|---|---|---|
| Синдром Халермана-Штрайфа (очно-щелепно-лицева дисцефалія) | Брахіцефалія, мікрогнатія, дзьобоподібний ніс, мікростомія, (пташиний профіль), прояви ектодермальної дисплазії, карликовий зріст, мікрофтальмія, катаракта, недорозвиток брів, вій, симптом голубих склер, ністагм | Не встановлена. Спорадичні випадки |
| Хромосоми 21-ї трисомії с-м (с-м Дауна) | Брахіцефалія, монголоїдний розріз очних щілин, епікант, дакріостеноз, блефарит, плями Брушвільда на райдужці, кератоконус і набряк рогівки, катаракта, страбізм, ністагм, виражені порушення рефракції | Хромосомний дизбаланс |
| Хромосоми 18 трисомія (с-м Едвардса) | Птоз, зменшення очних щілин, епікант, мікрофтальм, помутніння рогівки, анізокорія, катаракта, колобоми очного дна і дисків зорових нервів, єдина пупкова артерія, затримка психомоторного розвитку, множинні вади розвитку | Хромосомний дизбаланс |
| Хромосоми 13 трисомія (с-м Патау) | Мікроцефалія, полідактилія, розщілина губи та піднебіння, мікрофтальм, анофтальм, в деяких випадках циклопія, гіпоплазія райдужки, її синехії, помутніння рогівки, вр. катаракта, глаукома, дисплазія сітківки, колобоми райдужки, очного дна, утворення хряща в очному яблуці, гіпоплазія зорового нерва | Хромосомний дизбаланс |
| Хромосоми Х моносомії (с-м Тернера) | Набряк кистей та стоп у новонароджених, шкірні складки на шиї, ВВС, первинна аменорея, птоз, епікант, с-м голубих склер, катаракта, страбізм, ністагм | Хромосомний дизбаланс |
| Синдром котячого ока | Атрезія ануса, ВВС, вади нирок, преаурикулярні папіломи, колобома райдужної оболонки, очного дна, зорового нерва, антимонголоїдний розріз очей, епікант, мікрофтальм, катаракта, страбізм, ністагм | Спорадичні випадки. В деяких випадках мікроструктурна перебудова 22 хромосоми |
| Хвороба Фабрі (глікосфінголіпідліпідоз; дефіцит альфа-галактозидази) | Акропарестезії, ангіокератома, дистрофія рогівки, звивистісь і розширеність судин кон’юнктиви і сітківки, ниркова гіпертензія, набряк соска зорового нерва, катаракта (спицеподібні помутніння задньої поверхні капсули кришталика, помутніння переднього сектора кришталика) | Х зчеплений, рецесивний |
| Нетримання пігменту синдром (с-м Блоха-Сульцбергера) | Характерні зміни шкіри, аномалії зубів, катаракта, мікрофтальм, помутніння рогівки, атрофія зорового нерва | Х зчеплений домінантний |
| Гепатолентикулярної дегенерації с-м (хв. Вільсона) | Гепатоспленомегалія, ознаки порушення функції печінки, ЦНС, нирок, гіперкінези, псевдобульбарні розлади, кільце Кайзера-Флейшера на рогівці (накопичення пігменту, який містить мідь по периферії десцеметової оболонки), катаракта у вигляді соняшника, в деяких випадках – порушення акомодації | Аутосомно-рецесивний |
| Галактоземія (відсутність активності галактозо-1-фосфат-уріділтрансферази в еритроцитах) | Затримка психомоторного розвитку, гепатоспленомегалія, жовтяниця, діарея, анорексія, геморагічний синдром, катаракта | Аутосомно-рецесивний |
| Галактокінази дефіцит | Катаракта, інколи затримка розумового розвитку | Аутосомно-рецесивний |
| Галактозоепімерази дефіцит | Катаракта, затримка психомоторного розвитку, гепатоспленомегалія, жовтяниця, діарея | Аутосомно-рецесивний |
| Хондродисплазія точкова, ризомелічний тип | Низький зріст, розщелина тіл хребців, ризомелічне вкорочення кінцівок (плече, стегно), точкова мінералізація епіфізів, множинні контрактури суглобів. Мікроцефалія, двобічна катаракта, алопеція, іхтіозоформна дисплазія шкіри | Аутосомно-рецесивний |
| Ектодермальна дисплазія, гідротична | Дефіцит слізної рідини, який приводить до кератопатій, стеноз слізних отворів, катаракта, страбізм, рідке волосся, можлива алопеція, дистрофія нігтів, гіперкератоз долонь і стоп, гіперпігментація шкіри над суглобами та сосками | Аутосомно-домінантний |
| Іхтіоз-катаракта | Вроджений іхтіоз, кортикальна катаракта | Аутосомно-рецесивний |
| Ротмунда-Томсона синдром (с-м атрофічної пойкілодермії і катаракти) | Ушкодження шкіри – прогресуюча ерітема, телеангіектазії, рубці, атрофія, пігментація, депігментація, ламелярна катаракта, яка розвивається у віці 2-7 років, дистрофія рогівки, ознаки ектодермальної дисплазії, інколи гіпоплазія або відсутність І-го пальця, променева косорукість, гіпогеніталізм, розумова відсталість | Аутосомно-рецесивний |
| Окуло-церебро-ренальний синдром (с-м Лоу) | Вроджена катаракта в осіб чоловічої статі, дрібні ділянки помутніння кришталика у жінок-носіїв, глаукома, рідко – мікрофтальм, затримка зросту і психічного розвитку, м’язова гіпотонія, гіперрухливість суглобів, зниження глибоких сухожильних рефлексів, тубулопатія з генералізованою гіпераміноацидурією, протеїнурією | Х зчеплений, рецесивний |
| Фетальний синдром краснухи | Глухота сенсоневральна, катаракта (як правило, тверда, ядерна, перламутрова з відносно прозорим кортикальним кільцем), мікрофтальм, гіпоплазія райдужки, атрофія і спайки, вроджена глаукома, атрофія зорового нерва, ністагм, страбізм, ВВС. Поява симптомів залежить від гестаційного віку | Пренатальна краснушна інфекція |
| Катаракти-мікрокорнеа синдром | Мікрокорнеа, катаракта, міопія, колобома райдужної оболонки. Інколи глаукома, відшарування сітківки | Аутосомно-домінантний |
| Брахідактилія, адонтія, гіпотрихоз і альбінізм | Катаракта, міопія, ністагм, страбізм, дистихіаз, вкорочення метакарпальних та метатарзальних кісток, адонтія, брахідактилія, генералізований гіпотрихоз, гіпопігментація | Аутосомно-рецесивний |
| Окуло-денто-дигітальний синдром | Мікрофтальм, мікрокорнеа, вроджена катаракта, глаукома, гіпотелоризм, персистенція зіничної мембрани, атрофія зорового нерва, помутніння рогівки, епікант, неправильний ріст зубів, адонтія, вузький ніс з гіпоплазією крил, камптодактилія, синдактилія | Аутосомно-домінантний |
Номер з каталогу МІМ:
- 116200 Cataract 1, Multiple Types; CTRCT1
- 604307 Cataract 2, Multiple Types; CTRCT2
- 601547 Cataract 3, Multiple Types; CTRCT3
- 115700 Cataract 4, Multiple Types; CTRCT4
- 116800 Cataract 5, Multiple Types; CTRCT5
- 116600 Cataract 6, Multiple Types; CTRCT6
- 115660 Cataract 7, CTRCT7
- 115665 Cataract 8, Multiple Types; CTRCT8
- 604219 Cataract 9, Multiple Types; CTRCT9
- 600881 Cataract 10, Multiple Types; CTRCT10
- 610623 Cataract 11, Multiple Types; CTRCT11
- 611597 Cataract 12, Multiple Types; CTRCT12
- 116700 Cataract 13 with Adult I Phenotype; CTRCT13
- 601885 Cataract 14, Multiple Types; CTRCT14
- 615274 Cataract 15, Multiple Types; CTRCT15
- 613763 Cataract 16, Multiple Types; CTRCT16
- 611544 Cataract 17, Multiple Types; CTRCT17
- 610019 Cataract 18; CTRCT18
- 615277 Cataract 19, Multiple Types; CTRCT19
- 116100 Cataract 20, Multiple Types; CTRCT20
- 610202 Cataract 21, Multiple Types; CTRCT21
- 609741 Cataract 22, Multiple Types; CTRCT22
- 610425 Cataract 23, Multiple Types; CTRCT23
- 601202 Cataract 24; CTRCT24
- 605728 Cataract 25; CTRCT25
- 605749 Cataract 26, Multiple Types; CTRCT26
- 607304 Cataract 27; CTRCT27
- 609026 Cataract 28; CTRCT28
- 115800 Cataract 29; CTRCT29
- 116300 Cataract 30, Multiple Types; CTRCT30
- 605387 Cataract 31, Multiple Types; CTRCT31
- 115650 Cataract 32, Multiple Types; CTRCT32
- 611391 Cataract 33, Multiple Types; CTRCT33
- 612968 Cataract 34, Multiple Types; CTRCT34
- 609376 Cataract 35; CTRCT35
- 613887 Cataract 36; CTRCT36
- 614422 Cataract 37; CTRCT37
- 614691 Cataract 38; CTRCT38
- 615188 Cataract 39, Multiple Types; CTRCT39
- 302200 Cataract 40; CTRCT40
- 116400 Cataract 41; CTRCT41
- 115900 Cataract 42; CTRCT42
- 616279 Cataract 43; CTRCT43
- 616509 Cataract 44; CTRCT44
- 616851 Cataract 45; CTRCT45
- 212500 Cataract 46, Juvenile-Onset; CTRCT46
- 612018 Cataract 47; CTRCT47
Список літератури:
- Francois J. Congenital cataracts. Netherlands: C.C. Thomas, 1963.
- Francois J. Genetics of cataract. Ophtalmologia, 1982; 184: 61-71.
- Friedmann MW, Wright E.S: Hereditary microcornea and cataract in 5 generations. Am Ophtalmol, 1952; 35: 1017-1021.
- Maumenel I.H.: Classification of hereditary cataracts in children by linkage analysis. Frans Am Acad Ophtalmol Otolaryngol 1979; 86: 1554-1558.
- Nance WE, at al. Congenital X-linked cataract, dental anomalies and brachimetacarpalia. BD: OAS x (4). New York: March of Dimes Birth Defects Foundation, 1974: 285-291.
Переглянуто редакційною колегією I.B.I.S.: 15/02/2002
Глухота вроджена нейросенсорна і зоб
(Pendred Syndrome)
І.М. Дубчак
Лікар-генетик медико-генетичної консультації
Хмельницького пологового будинку
Синонім: синдром Пендреда.
Вперше описано Pendred у 1896 р. у двох сестер з зобом. Лише століття згодом Coyle et al. (1996) та Sheffield et al. (1996) дослідили, що ген хвороби знаходиться на хромосомі 7q.
Основні діагностичні критерії:
- Вроджена непрогресуюча нейросенсорна глухота;
- Еутиреоїдний зоб;
- Мальформація Мондіні або розширення вестибулярного каналу.
Клінічна діагностика:
- Зоб здебільшого дифузний, м’якої консистенції, але в деяких випадках спостерігаються вузли. Характерна еутиреоїдна струма, але можуть бути ознаки гіпотиреозу, особливо після тиреоїдектомії. В такому випадку у хворих спостерігається затримка зросту, надлишкова вага, хоча ожиріння рідко, блідість, анорексія, сухість шкіри, затримка статевого розвитку, брадікардія. Інколи у пацієнтів з еутиреоїдним зобом спостерігається надмірна відповідь на введення тиреотропного гормону, що свідчить про компенсований гіпотиреоїдизм. У понад 75% пацієнтів зоб виявляється при клінічному обстеженні. У 40% він розвивається у старшому дитячому та пубертатному віці, іноді в юнацькому, решта випадків припадають на дорослий вік. Гіперплазія залози більш виражена у жінок і пов’язана з порушенням перетворення йоду в органічну форму, що лабораторно може бути підтверджено перхлоратним тестом.
- Втрата слуху: ступінь вираженості втрати слуху може бути різною. Класично – це білатеральна виражена прелінгвальна нейросенсорна глухота. Причиною прогресивної втрати слуху є дисплазії завитка (cochlеa) та розширення вестибулярних каналів. Синдром Пендреда – єдине відоме генетичне захворювання, для якого характерний широкий вестибулярний канал. Вестибулярні порушення спостерігаються у 66% пацієнтів з синдромом Пендреда, коливаючись у широких межах від незначного однобічного ураження до повного випадіння функції. Недорозвинені перетинки у дистальних кільцях завитка (деформація Мондіні) виявились звичною, але не постійною ознакою синдрому Пендреда (у 85% випадків).
Діагностика:
- перхлоратний тест (при непорушеному процесі органіфікації йоду щитовидною залозою він становить менше 10%, у пацієнтів з синдромом Пендреда – значно зростає, сягаючи 15-80%). Перхлоратний тест є досить чутливим діагностичним тестом (псевдонегативні результати не перевищують 3%);
- визначення гормонів щитовидної залози (підвищення рівня тиреотропного гормону та зниження тироксину);
- консультація сурдолога;
- аудіометрія;
- томографія височних кісток;
- дослідження функцій вестибулярного апарату;
- ДНК-діагностика (виявлення мутації у гені SLC26A4 у локусі 7q22-q31) – позитивна у 75% випадків.
Диференційний діагноз:
Дефект транспорту йоду, інші асоціації глухоти з порушенням синтезу тироксину.
Етіологія:
У 75% пацієнтів виявляється дефект гена SLC26A4, локалізованого на довгому плечі 7 хромосоми (7q22-q31).
Описано 47 різноманітних мутацій гена SLC26A4, які є причиною синдрому Пендреда. Три мутації L236P, T416P, та 1001+1 Г на А є мажорними.
Нормальний генний продукт – ген, що кодує білок з 780 амінокислот (86 kDa), названий пендріном, який відповідає за перенос йоду та хлору.
Частота виникнення: 7 : 100000.
Batsakіs and Nіshіjama (1962) визначили, що синдром Пендреда складає від 1 до 10% усіх випадків спадкової глухоти. Варіабельна експресивність синдрому Пендреда, особливо його тиреоїдних проявів, може штучно занижувати реальну частоту захворювання.
Співвідношення статей: Ч1 : Ж1.
Тип успадкування: аутосомно-рецесивний.
Патогенез:
Мутація пендрін-гена (PDS-ген) призводить до порушення транспорту йоду і синтезу тироксину, що, в свою чергу, призводить до гіперплазії щитовидної залози і зобу. Роль зміни функції пендріну в зниженні слуху не досліджена. Мутований PDS-ген важко експресується, порушується сульфатація тироглобуліну. Вважають, що це може впливати на послідовне йодування білка. Роль пендріну у розвитку завитка є менш очевидною.
Вік маніфестації:
Нейросенсорна глухота проявляється з народження або в ранньому дитинстві. У 50% випадків дефект слуху різко виражений. Збільшення щитовидної залози спостерігається у віці 5-8 років, але в деяких випадках може бути і при народженні. У 60% дорослих з синдромом Пендреда є зоб.
Прогноз:
Сприятливий, якщо немає серйозних ускладнень і призначена раціональна терапія. Ризик малігнізації щитовидної залози мінімальний.
Лікування:
При еутиреоїдному зобі хворі знаходяться під спостереженням ендокринолога, при клінічних проявах гіпотиреозу проводиться замісна терапія гормонами щитовидної залози. Тканина щитовидної залози дуже добре регенерує. Тому при гіперплазії щитовидної залози показане хірургічне втручання (резекція).
Порушення слуху коригується сурдологом, зазвичай, використовуються слухові апарати, у деяких випадках – імплантація завитка. Хворі з синдромом Пендреда потребують постійного спостереження педіатра, ендокринолога, сурдолога.
Нагляд:
- Освітні програми;
- Регулярний огляд (щопівроку) лікарем пробанда та його родичів;
- Регулярний огляд (щопівроку) ендокринологом пацієнта з синдромом Пендреда та його найближчих родичів;
- Повторна аудіометрія;
- При потребі – імплантація завитка.
Запобігання:
Генетичне консультування може виявити хворих із синдромом Пендреда серед сибсів. Оскільки повторний ризик захворювання у сім`ї з ураженим пробандом є високим (25%), то рекомендується проведення пренатальної ДНК-діагностики шляхом біопсії хоріона на 10-12 тижні вагітності.
Важливим є запобігання виникнення ускладнень у хворих із синдромом Пендреда.
Номер з каталогу МІМ:
274600 Pendred Syndrome; PDS.
Література:
- Козлова С.И., Демикова Н.С., Селанова О., Блинникова О.Е. Наследственные синдромы и медико- генетическое консультирование.- М.: Практика, 1996.
- Bargman, G. J.; Gardner, L. I. Otic lesions and congenital hypothyroidism in the developing chick. J. Clin. Invest. 46: 1828-1839, 1967.
- Coyle, B.; Coffey, R.; Armour, J. A. L.; Gausden, E.; Hochberg, Z.; Grossman, E.; Britton, K.; Pembrey, M.; Reardon, W.; Trembath, R. Pendred syndrome (goiter and sensorineural hearing loss) maps to chromosome 7 in the region containing the nonsyndromic deafness gene DFNB4. Nature Genet. 12: 421-423, 1996.
- Cremers, C. W. R. J.; Bolder, C.; Admiraal, R. J. C.; Everett, L. A.; Joosten, F. B. M.; van Hauwe, P.; Green, E. D.; Otten, B. J. :Progressive sensorineural hearing loss and a widened vestibular aqueduct in Pendred syndrome. Arch. Otolaryng. Head Neck Surg. 124: 501-505, 1998.
- Elman, D. S. : Familial association of nerve deafness with nodular goiter and thyroid carcinoma. New Eng. J. Med. 259: 219-223, 1958.
- Everett, L. A.; Belyantseva, I. A.; Noben-Trauth, K.; Cantos, R.; Chen, A.; Thakkar, S. I.; Hoogstraten-Miller, S. L.; Kachar, B.; Wu, D. K.; Green, E. D. : Targeted disruption of mouse Pds provides insight about the inner-ear defects encountered in Pendred syndrome. Hum. Molec. Genet. 10: 153-161, 2001.
- Fraser, G. R. :Association of congenital deafness with goiter (Pendred’s syndrome a study of 207 families.): Ann. Hum. Genet. 28: 201-249, 1965.
- Gomez-Pan, A.; Evered, D. C.; Hall, R. : Pituitary-thyroid function in Pendred syndrome. Brit. Med. J. 2: 152-153, 1974.
- Hollander, C. S.; Prout, T. E.; Rienhoff, M.; Ruben, R. J.; Asper, S. P., Jr. : Congenital deafness and goiter: studies of a patient with a cochlear defect and inadequate formation of iodothyronines. Am. J. Med. 37: 630-637, 1964.
- Illum, P.; Kiaer, H. W.; Hvidberg-Hansen, J.; Sondergaard, G. : Fifteen cases of Pendred’s syndrome. Arch. Otolaryng. 96: 297-304, 1972.
- Kopp, P.; Karamanoglu Arseven, O.; Sabacan, L.; Kotlar, T.; Dupuis, J.; Cavaliere, H.; Santos, C. L. S.; Jameson, J. L.; Medeiros-Neto, G. : Phenocopies for deafness and goiter development in a large inbred Brazilian kindred with Pendred’s syndrome associated with a novel mutation in the PDS gene. J. Clin. Endocr. Metab. 84: 336-341, 1999.
- Masmoudi, S.; Charfedine, I.; Hmani, M.; Grati, M.; Ghorbel, A. M.; Elgaied-Boulila, A.; Drira, M.; Hardelin, J.-P.; Ayadi, M. : Pendred syndrome: phenotypic variability in two families carrying the same PDS missense mutation. Am. J. Med. Genet. 90: 38-44, 2000.
- Medeiros-Neto, G. A.; Nicolau, W.; Kieffer, J.; Ulhoa Cintra, A. B. : Thyroidal iodoproteins in Pendred’s syndrome. J. Clin. Endocr. 28: 1205-1213, 1968.
- Milutinovic, P. S.; Stanbury, J. B.; Wicken, J. V. : Thyroid function in a family with the Pendred syndrome. J. Clin. Endocr. 29: 962-969, 1969.
- Pendred, V. : Deaf-mutism and goitre. Lancet II: 532, 1896.
- Phelps, P. D.; Coffey, R. A.; Trembath, R. C.; Luxon, L. M.; Grossman, A. B.; Britton, K. E.; Kendall-Taylor, P.; Graham, J. M.; Cadge, B. C.; Stephens, S. G. D.; Pembrey, M. E.; Reardon, W. : Radiological malformations of the ear in Pendred syndrome. Clin. Radiol. 53: 268-273, 1998.
- A report of the occurrence of deaf-mutism and goiter in four of six siblings of a North American family. Ann. Surg. 146: 941-948, 1957.
Переглянуто редакційною колегією I.B.I.S.: 29/06/2003
Вроджена дисплазія кульшових суглобів
(Congenital Hip Dysplasia)
Е. Пшенична
Лікар-травматолог
Рівненської обласної дитячої клінічної лікарні
Вроджена дисплазія кульшового суглоба (ДКС) – найбільш розповсюджений вид деформації опорно-рухового апарату у дітей. Ця патологія характеризується вадою формування всіх частин суглоба (ацетабулярної западини, голівки стегнової кістки, прилеглих м’язів, сумково-зв’язкового апарату), а також пошкодженням або недорозвитком його окремих елементів.
Термін “дисплазія кульшового суглоба” був вперше запропонований M. Schede (1900) для визначення стану, який передує вродженому вивиху стегна і характеризується недорозвитком тканин тазостегнового суглоба.
Для визначення недорозвитку кульшового суглоба користуються терміном “прелюксація”, який введено у вітчизняну літературу В.О. Марксом (1934). Застосовуються також терміни La hanche luxante, Luxationschufte, Malformations luxantes la hanche, Displacement of the hip Somerville.
За даними В.І. Овваді, В. Фрейка, дисплазія кульшових суглобів виявляється у 10-20% новонароджених. Часто порушення формування кульшових суглобів поєднується з загальним недорозвитком дитини, про що свідчить те, що у недоношених дітей вроджена дисплазія розвивається в 10 разів частіше, ніж у доношених. Співвідношення дівчат і хлопчиків з ДКС у віці до 1 року складає 3,1:1.
Схильність до дисплазії та вивих стегна за даними Я.Б. Куценка, Е.Ф. Лордкіпанідзе, О.П. Меженіної успадковується в основному за полігенним типом, а утворення вродженого вивиху стегна залежить від багатьох ендогенних та екзогенних факторів. Під час обстеження мають значення наявність у родичів не тільки вродженого вивиху стегна, але й інших захворювань кульшового суглоба, виникнення яких часто пов’язане з його недорозвитком (хвороба Пертеса, coxa vara, coax valga, остеоартрит і ін.), а також наявність “сімейного” розслаблення капсули суглобів.
Прогноз:
З питання про причини розвитку дисплазії кульшових суглобів існує концепція про “відносну незрілість тканин і диспропорцію дозрівання”, які можуть бути причиною розвитку патологічних станів. Поняття “дисплазія” охоплює всі можливі випадки неправильного росту і розвитку кульшового суглоба.
В якості етіологічних факторів, які призводять до відхилень в нормальному розвитку кульшового суглоба і оточуючих м’язів, називають ваду їх первинної закладки, затримку розвитку кульшового суглоба під час внутрішньоутробного життя плоду. Це підтверджується спостереженнями порушень метаболізму, гіпоксії, тиреотоксикозу, з гормональної недостатності у жінок, які народили дітей з вродженою дисплазію. У 1/3 з них були виявлені ознаки універсального геніального інфантилізму.
Механічна теорія виникнення дисплазії пояснює розвиток цієї патології неправильною позицією плода в матці, підвищеним тонусом її стінок, маловіддям. Для перетворення хряща в кістку як в плідний період, так і після пологів необхідний взаємний тиск суглобних кінців. Будь – який фактор, що зменшує його, викликає порушення осі фіксації, заважає нормальному розвитку кульшових суглобів і призводить до дисплазії.
Великий вплив мають механічні фактори і на розвиток кульшового суглоба в постанатальний період. Правильне сповивання і догляд за новонародженими є ефективними заходами, які сприяють профілактиці і лікуванню вродженої дисплазії кульшових суглобів. Тісне сповивання порушує розвиток кульшового суглоба, сприяє прогресуванню дисплазії і утворенню підвивиху і вивиху стегна.
В розвитку дисплазії важливу роль відіграє розслаблення капсули і зв’язок кульшового суглоба. Основною структурою, яка забезпечує стабільність кульшового суглоба у плодів і дітей до 1 року, є сумково – зв’язковий апарат. При його слабкості, у тому числі обумовленій конституційно, схильність до розвитку дисплазії і вивиху стегна більш виражена.
Разом з теорією сумково – зв’язкової релаксації існує також гормональна теорія. Згідно цієї теорії, причиною розвитку дисплазії є слабкість капсули кульшового суглоба. При гіпофункції щитовидної залози порушується розвиток сполучної тканини, спостерігається енходральне окостеніння, при сповільненому розвитку кульшового суглоба можуть виникати дисплазія та вивих стегна.
Патогенез:
Дисплазія кульшового суглоба характеризується гіпоплазією ацетабулярної западини, її плескатістю, малими розмірами голівки стегна і сповільненням її окостеніння, поворотом верхнього кінця стегна до переду (антеторсією), аномаліями в розвитку нервово – м’язового апарату ділянки тазостегнового суглоба. Ці зміни підтверджуються патологоанатомічними даними: головка стегна в перші місяці життя дитини зміщена назовні і незначно догори. Вертлюжна западина не тільки плеската, але й витягнута в довжину, її верхньо – задній край недорозвинутий, внаслідок чого голівка скошена і зверху відсутній кістковий упор для голівки стегна. Плескатість ацетабулярної западини збільшується ще за рахунок потовщення хрящового шару дна западини і розвитку на її дні сполучної тканини. Головка стегна меншого розміру, деформована, з’являється пізніше.
Діагностика і клініка:
В групу дисплазій кульшового суглоба у дітей раннього віку входять такі захворювання, як: вроджений передвивих, вроджений підвивих, вроджений вивих і рентгенологічно незрілий кульшовий суглоб. Найбільш простою організаційною формою раннього виявлення вроджених дисплазій є систематичний огляд всіх новонароджених в пологовому будинку. Незважаючи на те, що клініка дисплазій в перші дні життя дитини вкрай бідна на симптоми, вона достатня для того, щоб при певних навичках своєчасно діагностувати або запідозрити це захворювання.
Для правильної оцінки результатів клінічного обстеження огляд новонародженого необхідно проводити за спеціальною методикою.
Найбільш частими симптомами вроджених дисплазій кульшового суглоба є:
- Oбмеження відведення в кульшових суглобах.
- Симптом ковзання або вправлення і вивихування (симптом Маркса-Ортолані).
- Асиметрія складок на стегні і сідничних складок.
- Вкорочення нижньої кінцівки, яке визначається візуально.
- Зовнішня ротація нижньої кінцівки.
Ці симптоми треба шукати і знаходити, так як вони не завжди достатньо ясно визначаються і для їх розпізнавання потрібні певні навички в обстеженні дитини.
- У дитини, яка лежить на спині, спостерігається обмеження пасивного відведення ніг, зігнутих під прямим кутом в кульшових і колінних суглобах. Це найбільш рання і постійна ознака вродженої патології. Обмеження відведення з часом наростає. При нормальних кульшових суглобах відведення стегон буде майже повним; при наявності дисплазії завжди має місце цей симптом. Обмеження відведення стегон можливе при спастичному паралічі, м’язовій контрактурі, яка спостерігається у новонароджених і в інших суглобах, а також при вродженій варусній деформації шийки стегна. Всі ці захворювання повинні бути виключеними шляхом вивчення стану всіх м’язів і за допомогою рентгенограми кульшових суглобів. Важливо знати, що фізіологічна ригідність м’язів новонародженого не буває постійною. В певні моменти вдається відвести стегна, чого не буває при дисплазія до вправлення голівки.
- Дуже важливим, найбільш раннім, але не постійним є симптом ковзання, який описаний вперше в 1925 році Hilgenreiner. Його також називають симптомом Маркса – Ортолані, симптомом нестійкості. Суть симптому полягає в тому, що при відведенні ніжок відбувається вправлення вивиху, яке супроводжується лусканням і здригуванням ніжки. Для виявлення цього симптому існує спеціальна методика обстеження новонародженого, при якій лікар, згинаючи обидві ніжки в кульшовому і колінному суглобах, великі пальці розташовує на внутрішніх, а інші пальні – на зовнішніх поверхнях стегон. Повільно, уникаючи форсованих рухів, лікар відводить стегна рівномірно в обидва боки. Симптом Маркса – Ортолані, як правило, зникає до 5-7 днів життя дитини, але у деяких дітей при наявності м’язової гіпотонії може зберігатись протягом перших місяців життя.
- Асиметрія складок на стегні або нерівномірна їх кількість може також свідчити про наявність дисплазії. На боці дисплазії складок більше, вони є більш глибокими, ніж на здоровому боці і розташовані проксимальніше. Симптом цей не є абсолютним і окремо, без інших даних, не може прийматись до уваги, так як спостерігається лише у 2/3 хворих і може зустрічатись у здорових дітей. При огляді сідничні складки можуть бути не на одному рівні. Цей симптом характерний для одностороннього вивиху стегна. Крім того, у здорових дітей між стегнами і тулубом ззаду є глибокі симетричні складки. Асиметрія цих складок або їх відсутність свідчать про наявність одно – або двобічного вивиху.
- Одним із симптомів дисплазії кульшового суглоба може бути зовнішня ротація ноги на боці вивиху. Вона добре визначається, коли дитина спить – на цей симптом звертають увагу самі матері.
- Вкорочення нижньої кінцівки, яке визначається візуально, характерне для високих вивихів. Воно може спостерігатись не тільки при явному односторонньому вивиху, а і при різних дисплазіяч, навіть двобічних, але з різним розташуванням стегон по висоті. Визначити довжину і вкорочення ніг у грудних дітей сантиметровою стрічкою важко. Про різницю в довжині ніг судять по розташуванню рівнів колінних суглобів, зігнутих і приведених до живота.
Всі перелічені симптоми можуть спостерігатись разом або може мати місце частина симптомів, в останньому випадку краще запідозрити вроджену патологію кульшового суглобу і провести рентгенографію. Запідозрений, але не підтверджений вивих стегна, свідчить лише про уважність лікаря і шкоди дитині не завдасть. Пропущене ж захворювання може зробити дитину важким інвалідом на ціле життя.
Діагностика:
Рентгенологічному методу дослідження належить значна роль в діагностиці дисплазії кульшових суглобів у новонароджених. Під час рентгенографії дитина лежить на спині з витягнутими і приведеними ногами в положенні незначної ротації, суворо симетрично, таз повинен щільно прилягати до касети. Необхідний захист статевих органів свинцевою пластинкою, яка при правильному її положенні не заважає рентгенографії.
При рентгенодіагностиці захворювань кульшових суглобів слід враховувати, що у новонароджених відсутні ядра окостеніння головок стегон і необхідно пам’ятати, що висота голівки стегна дорівнює ширині шийки стегна. Ацетабулярна западина також є хрящовою і не дає конкретної тіні при читанні рентгенограми. Особливого значення надається стану верхнього краю ацетабулярної западини. Важливо врахувати також розташування хрящової голівки – наскільки вона вище і латеральніше за своє нормальне положення.
При наявності вродженого вивиху стегна відмічається косе розташування верхнього краю ацетабулярної западини, а верхній кінець стегна, і також передбачена, але ще не видима на рентгенограмі хрящова голівка стегна, знаходяться більше латерально, в деяких випадках (навіть якщо дитина ще не стоїть) вище на здоровому боці. Природно, що при двобічному вивиху рентгенодіагностика буває важкою через неможливість провести порівняння із здоровим суглобом. В таких випадках застосовуються спеціальні схеми, запропоновані Омбреданом, Хильгенрейнером, Ерлахером, Путті, Рейнбергом. Такі схеми певними лініями визначають нормальне розташування елементів кульшового суглобу і відповідно дозволяють визначити ступінь цього зміщення. Ранні рентгенологічні симптоми вродженого вивиху стегна були вперше визначені болонським ортопедом Путті. Він запропонував класичну “тріаду Путті”:
- підвищена скошеність дахівки ацетабулярної западини;
- зміщення проксимального кінця стегна назовні і догори відносно ацетабулярної западини;
- пізня поява і гіпоплазія ядра окостеніння.
Так як ядро окостеніння голівки стегна з’являється в нормі в 4-6 місячному віці, а при дисплазії – пізніше, до 9-10 місяця, то у дітей перших місяців життя, коли голівка, крім того, знаходиться лише латерально, але не вище западини, раціонально використовувати для оцінки рентгенограми схему Хільгенрейнера. Проводиться горизонтальна лінія через обидва У – подібні хрящі (лінія Келлера); від найбільш високо розташованого рівня діафізу стегна проводять перпендикуляр h до перетинання з горизонтальною лінією. Відстань від основи У-подібного хряща до перпендикуляра h називається величиною d. Довжина горизонтального відрізка d в нормі дорівнює 1,2-1,2 см, так само, як і довжина перпендикуляра h. При вивиху величина h зменшується, а d – збільшується. Від дна ацетабулярної западини проводиться лінія, яка торкається до найбільш периферичного відділу дахівки западини. Кут, який утворюється (індекс) в нормі у новонародженого дорівнює 27-30°, у дворічної дитини наближається до 20°. При наявності дисплазій суглобу і відставання в розвитку ацетабулярної западини індекс збільшується. Для дисплазії кульшових суглобів характерним є не тільки абсолютне збільшення цього кута, скільки наявність різниці в кутах з обох боків, що яскраво свідчить про ваду розвитку тазу.
Схема, запропонована С.А. Рейнбергом, застосовується у дітей більш старшого віку. Проводяться три вертикальні лінії: по середній лінії тіла, через верхньолатеральний край (виступ дахівки) здорової ацетабулярної западини і на рівній відстані від середньої лінії на боці вивиху. В нормі вертикальна лінія проходить через діафіз стегна, а при вивиху – медіальніше. Горизонтальна лінія також проводиться через У – подібні хрящі. В нормі голівка стегна розташована медіальніше вертикальної і нижче горизонтальної лінії, при вивиху – латеральніше і вище вказаних ліній. У дітей другого півріччя життя звертають увагу на лінію Шентона. В нормі нижній контур шийки стегна переходить в верхню півокружність запирального отвору, а при вивиху дугоподібна лінія відсутня, так як з’являється уступ через більш високе розташування нижнього контуру шийки стегна.
Найбільшу розповсюдженість отримала схема Омбредана, де враховуються положення голівки по відношенню до горизонтальної лінії (лінії Келлера) і розташування діафізу стегна (лінії Омбредана) по відношенню до вертикальної лінії.
Таким чином, певні навички дозволяють дати клінічно-рентгенологічну оцінку стану кульшових суглобів у дітей перших тижнів життя.
Ультразвукове дослідження:
Новим методом, який розширює діагностичні можливості оцінки стану розвитку кульшового суглоба у новонароджених дітей та дітей у віці до 6-8 місяців, є ультразвукове дослідження, що дає можливість ідентифікувати м’якотканинні компоненти кульшового суглоба – неосифіковану ацетабулярну западину, голівку стегнової кістки, У-подібний хрящ, лімбус, зв’язку голівки стегнової кістки, капсулу кульшового суглоба і м’язи, визначити співвідношення суглобових кінців, виявити порушення їх розвитку. Дослідження проводиться на електронному сканері з частотою сигналу 5 мГц (у новонароджених 7,5 мГц) в режимі реального часу в положенні дитини на боку. Датчик ставлять суворо перпендикулярно латеральній поверхні кульшового суглобу і паралельно осі стегнової кістки. Стегно згинається під кутом 10-20° і злегка ротується всередину. На основі даних ультразвукової діагностики виділяються групи дітей: діти, які не потребують лікування і контролю (1 тип за Графом); діти, які потребують контролю (2 тип за Графом); діти, які потребують лікування (3-4 типи). Таким чином, ультразвукове дослідження новонароджених і дітей у віці 1-6 місяців дозволяє визначити надійні критерії, які характеризують розвиток кульшових суглобів. Застосування цього методу особливо раціональне при необхідності уточнити діагноз у дітей в віці до 3 місяців, у яких виявлені фактори ризику або клінічні ознаки дисплазій кульшових суглобів.
Диференційний діагноз:
Дисплазії кульшових суглобів у грудних дітей повинні розмежовуватись тільки з вродженим вкороченням стегна або іншою рідкою аномалією розвитку (повний недорозвиток проксимального кінця стегнової кістки), а також з фізіологічною або спастичною м’язовою гіпертонією, яка зникає або проходить після деякого періоду на тяжіння м’язів.
Паралітичний вивих стегна після поліомієліту супроводжується характерним анамнезом, атрофією сідничних і стегнових м’язів і на рентгенограмі відрізняється остеопорозом кісток без плескатості ацетабулярної западини.
Патологічний вивих стегна після епіфізарного остеомієліту спостерігається тільки у грудних дітей внаслідок накопичення гною, а частіше реактивної рідини в кульшовому суглобі. Висока септична температура в анамнезі, наявність рубців від нориць, можливі деструктивні зміни голівки характерні для цього захворювання.
Консервативне лікування дисплазії стегна:
Лікування ДКС найбільш наочно ілюструє необхідність дотримання всіх принципів дитячої ортопедії: діагностувати захворювання потрібно з народження, розпочати лікування необхідно в пологовому будинку або в перші тижні життя дитини, консервативне лікування передує можливому оперативному лікуванню, після закінчення лікування необхідна профілактика рецидивів. З кожним місяцем життя дитини ускладнюються методи лікування захворювання і погіршується функціональний результат запізнілої терапії.
Консервативне лікування ДКС бажано почати в пологовому будинку. Пеленати тісно не слід, ніжки повинні лежати вільно. Бажано, щоб перші дні, до отримання відвідної шини, дитина лежала на спині з розведеними ногами. Для цього між ними прокладають пелюшку, а під нею – між зігнутими і відведеними колінними суглобами кладуть невелику подушечку з дитячої церати. Після рентгенологічного дослідження вирішується питання про метод лікування дисплазії. Якщо має місце тільки вроджений передвивих, то на прямому знімку кульшових суглобів проксимальний кінець стегна знаходиться дещо латеральніше за норму, а на знімку з відведенням ніг він наближається до западини. Таким дітям проводиться лікування на шині з розігнутими, але відведеними ногами. При вродженому вивиху стегна, який має тератологічний характер, тобто вивих вже сформований до початку стояння дитини, на прямому рентгенівському знімку проксимальний кінець стегна знаходиться також дещо латеральніше вертлюжної западини, але може бути на її рівні або дещо вище. На знімку у відведенні проксимальний кінець стегна не входить в западину, а виходить вище. Таким дітям вже з народження безумовно треба застосовувати шини, які надають зігнутим в колінних суглобах ногам положення відведення.
В ортопедії прийнято визначати положення згинання ніг під прямим кутом в кульшових і колінних суглобах і повного відведення стегон до площини ліжка, як положення Лоренц І. Дещо випростане положення ніг до тупого кута у вказаних суглобах носить назву положення Лоренц ІІ, а повністю випростані, але дещо відведені ноги – положення Лоренц ІІІ.
Для лікування вродженого передвивиху в положенні Лоренц ІІІ застосовуються спеціальні відвідні шини Віленського – розведення ніг досягається металевою рухомою розпіркою, яка фіксується шкіряними манжетками в ділянці нижньої третини гомілок.
При лікуванні справжніх вивихів або виражених дисплазій застосовуються різного роду шини в положенні Лоренц І. Зручна в усіх випадках вже в пологовому будинку шина – ліф ЦІТО.
Дитина знаходиться в шині протягом всього часу доби терміном 4 місяці і, якщо відмічається формування більш глибокої ацетабулярної западини (на основі вимірювання ацетабулярного кута), шину знімають. В рідких випадках лікування на шині продовжується до 6 місяців.
Термін лікування найбільш легких дисплазій на розпорці Віленського – 3 місяці. Крім того, можуть бути застосовані інші аналогічні засоби: шина Розена, пластмасова шина Волкова, абдукційна шина Шнейдерова.
Паралельно із вказаними шинами у дітей до 3 місяців можуть застосовуватись різні м’які пристосування, які створюють необхідну правильну укладку хворого: ліфчик Байера, стремена Павлика та інші.
При етапному лікуванні дітей з вродженою патологією кульшового суглоба майже у 50% випадків спостерігається симптоми рахіту, запізніле прорізування зубів, скошеність потиличних кісток та ін. У дітей з ознаками рахіту процеси формування ацетабулярної западини відбуваються більш сповільненими темпами. Необхідно проводити енергійну протирахітну терапію: цикли вітамінотерапії, збагачення організму дитини солями – перевід на овочевий прикорм, призначення глюконату кальцію, ультрафіолетове опромінення. Система заходів з профілактики рахіту у дітей повинна входити як обов’язкова частина етапного лікування вроджених дисплазій кульшових суглобів.
Профілактика дисплазій кульшових суглобів:
Всі заходи, які проводяться акушерами-гінекологами по попередженню і лікуванню патології вагітності можна вважати профілактичними в плані попередження порушень розвитку кульшового суглобу.
Найбільше значення має післяпологова профілактика порушень розвитку кульшового суглобу. В цей період ортопеди можуть і повинні рекомендувати заходи, що сприяють правильному формуванню кульшового суглобу.
Необхідно проводити аналіз частоти виявлення вроджених деформацій опорно-рухового апарату в кожному пологовому будинку. Дуже важливий в цьому плані взаємний зворотній зв’язок пологового будинку і дитячої поліклініки. Списки всіх дітей з виявленою в пологовому будинку ортопедичною патологією необхідно щомісяця передавати в дитячу поліклініку за місцем проживання дитини. За кожним пологовим будинком повинен бути закріплений дитячий ортопед. Оптимальним є відвідування пологового будинку ортопедом 2 рази на тиждень.
Для попередження постнатального виникнення і прогресування дисплазій надзвичайно важливим є навчання матерів правильному повиванню новонароджених, ознайомлення їх з ранніми ознаками вродженої дисплазії кульшових суглобів.
Широке (вільне) повивання сприяє нормальному формуванню кульшового суглоба і попереджає розвиток підвивиху або вивиху стегна. В перші тижні і місяці життя широке повивання корисне всім дітям, але особливо тим, у яких є фактори ризику порушення кульшових суглобів (недоношеним, “штучникам”, гіпотрофікам, дітям, які народились в сідничному передлежанні тощо).
Диспансерний нагляд за хворими на дисплазію кульшових суглобів:
- Огляд ортопеда в пологовому будинку.
- Огляд в 1 місяць – при першому відвідуванні дитиною дитячої клініки.
- Огляд в 3 місяці, коли вже можна проводити рентгенограму кульшових суглобів.
- Огляд в 11-12 місяців, коли дитина починає стояти і ходити.
Номер з каталогу МІМ:
142700 Developmental Dysplasia of the Hip 1; DDH1.
Література:
- Альбамасова Е.А., Лузина Е.В. Развитие тазобедренного сустава после лечения врожденного подвывиха и вывиха бедра у детей. – Ташкент: Медицина,1983.- С. 183.
- Волков М.В., Тер-Егизаров Г.М., Юкина Г.П. Врожденный вывих бедра.- В кн.: Ортопедия и травматология детского возраста.- М.: Медицина,1983.- С. 129-158.
- Куценюк Я.Б., Рулла Э.А., Мельник В.В. Врожденная дисплазия тазобедренного сустава. Врожденные подвывих и вывих бедра. – Киев. Здоров’я, 1992, стр. 184.
- Садофьева В.А. Рентгено-функциональная диагностика заболеваний опорно-двигательного аппарата у детей. – М.: Медицина,1986.- С. 239.
- Соколовский А.М. К вопросу восстановления конгруэнтности при дисплазии тазобедренного сустава //Ортопедия, травматология и протезирование.- 1982.- №5.- С. 34-38.
- Стаматин С.И., Морару А.Т. Диагностика и лечение врожденного вывиха бедра. – Кишинев: Штиинца,1986.- С. 156.
- Фищенко П.Я., Хрулева К.Н., Оноприенко А.А. Раннее выявление и лечение остаточной нестабильности тазобедренного сустава после консервативного вправления врожденного вывиха бедра у детей: Методические рекомендации. – М.,1982.- С. 25.
- Шумада И.В. Раннее выявление и лечение врожденного вывиха бедра у детей //Ортопедия, травматология и протезирование. – 1988.- №18.- С. 3-8.
Переглянуто редакційною колегією I.B.I.S.: 11/06/2002
Вроджена глаукома
(Congenital Glaucoma)
І.П. Погребняк
Лікар-педіатр
Рівненської міської дитячої лікарні
Включення:
Вроджений буфтальм, вроджений гідрофтальм.
Основні діагностичні критерії:
Підвищений внутрішньоочний тиск, збільшення рогівки, збільшення очного яблука, витончення склери, зниження зорових функцій ока, екскавація диска зорового нерва, розширення передніх міліарних судин, розриви десцеметової оболонки.
Деякі статистичні дані про вроджену глаукому:
Вроджена глаукома була відома ще Гіппократу, Цельсію та Галену, але тоді це захворювання не було відокремлене від екзофтальму. Перша згадка про підвищений внутрішньоочний тиск у дітей належить Boerhaave (1749).
Вроджена глаукома або гідрофтальм зустрічається відносно рідко (1 випадок на 5000-10000 новонароджених), але в структурі вроджених дефектів органів зору їй належить досить відчутне місце – 3,5-6%.
На думку Е.І. Ковалевського (1986), в 90% випадків ця патологія може бути діагностована в пологовому будинку, але реалізувати цей показник на практиці доволі важко.
У 40-60% дітей захворювання проявляється при народженні чи на 1-му місяці життя, у 60-80% – в першому півріччі, а в 80-90% на першому році життя. Дане захворювання є однією з основних причин сліпоти у дітей, становлячи від 2,4% до 13,5% всіх випадків сліпоти у дітей.
Клінічна картина:
Ранні клінічні прояви включають сльозоточивість, світлобоязнь, блефароспазм, помутніння рогівки (набряк), розриви десцеметової оболонки, екскавацію та атрофію диска зорового нерва. Очне яблуко збільшене. Патологія є двобічною у 75% випадків і частіше зустрічається у хлопчиків.
Численні клінічні дослідження показують, що вроджена глаукома може з’являтись до моменту народження дитини.
При вродженій глаукомі дитина народжується з великими “виразними” очима. Характерним є збільшення розмірів очей відразу після народження, при цьому воно швидко прогресує.
Клінічна класифікація (за даними Є.Г. Сидорова, М.Г. Мирзаянца) – вроджена глаукома класифікується за такими ознаками:
-
- Форма;
- Патологія райдужко-рогівкового кута;
- Стадія;
- Внутрішньоочний тиск;
- Перебіг;
- Динаміка;
- Виникнення.
Форма захворювання:
-
- Проста (лише з аномаліями розвитку дренажної ділянки ока);
- З супутньою місцевою патологією (анірідія, колобома райдужки, синдром Франка-Каменецького та ін.);
- З супутньою місцевою і/або загальною патологією (синдроми Штурге-Вебера, Ригера, Лоу, Дауна, нейрофіброматоз та ін.)
Патологія райдужко-рогівкового кута:
-
- Гоніодисгенез І ступеня (кут відкритий, видно війчасте тіло);
- Гоніодисгенез ІІ ступеня (прикріплення райдужки на рівні задньої третини трабекули);
- Гоніодисгенез ІІІ ступеня (прикріплення райдужки на рівні середини трабекули чи більше спереду).
Стадія:
-
- Початкова: ДР (діаметр рогівки) більше, ніж на 1-2 мм від норми, ГК (глибина камери) – на 0,5 мм, ПЗВ (передньо-задня вісь ока) – на 1-2 мм, Е/Д (співвідношення горизонтальних діаметрів екскавації та диска зорового нерва) – до 0,3 мм;
- Розвитку: ДР збільшений на 2,3-3,5 мм, ГК – на 1-1,5 мм, ПЗВ – на 2,5-5 мм, Е/Д = 0,3-0,5 мм;
- Прогресуюча: ДР збільшений на 4 мм, ГК – на 2 мм, ПЗВ – на 5,5 мм і більше, Е/Д – 0,55 мм;
- Термінальна: грубі анатомічні зміни ока, буфтальм, атрофія зорового нерва, залишковий зір чи сліпота.
Внутрішньоочний тиск:
-
- Нормальний (до 22 мм рт. ст.);
- Помірно підвищений (23-30 мм рт. ст.);
- Високий (31-40 мм рт. ст.);
- Дуже високий (більше 41 мм рт.ст.).
Перебіг:
-
- Злоякісний;
- Типовий;
- Доброякісний;
- Абортивний (спонтанна зупинка прогресування).
Динаміка:
-
- Повне зникнення синдрому рогівки;
- Зникнення глаукоматозної екскавації;
- Нормалізація офтальмотонусу;
- Відсутність патологічного збільшення ока;
- Стабільний стан зорових функцій.
Виникнення:
-
- Спадкове;
- Внутрішньоутробне.
Асоційовані вади:
Серед них: анірідія, катаракта, глухота, розумова відсталість, патологія серця, синдром Марфана, синдром П’єра-Робена, гомоцистінурія, синдром Аксенфельда, синдром Лоу, сферофакія, синдром Штурге, нейрофіброматоз, трисомія D, трисомія F, синдром Рубінштейна-Тейбі, фетальний синдром краснухи (Грега).
Зупинимося на найбільш частих захворюваннях та синдромах, поєднаних з вродженою глаукомою.
Анірідія – як правило, двобічна генетично обумовлена вроджена аномалія, що передається по аутосомно-домінантному типу з високою пенетрантністю. Відсутність райдужки ніколи не буває повною. Інколи спостерігається підвивих і вивих кришталиків. Зір знижений. Наявний ністагм, косоокість. Зустрічаються полідактилія, дизостози черепа та обличчя, деформації кінцівок, вушних раковин, гідроцефалія, розумова відсталість. Глаукома наявна в половині випадків хворих з анірідією.
Синдром Франка-Каменецького. Наявний тільки у чоловіків, передається по рецесивному, зчепленому з Х-хромосомою типу. Жінки-носії передають мутантний ген половині своїх дочок та синів. Ризик носійства для дочок та захворювання для синів складає 50%.
Характерна двобічна різка гіпо- чи аплазія строми райдужки. Зона зіниці виглядає як сіро-голубе чи жовте кільце, війчаста зона має шоколадний колір. Часті аномалії зіниці (дистопія, неправильна форма, відсутність пігментної кайми).
Глаукома розвивається в другій декаді життя (хоча може бути і в грудному віці).
Синдром Ригера (мезодермальний дизгенез райдужки та рогівки). Діагноз встановлюється за трьома ознаками: часткова або повна аплазія переднього стромального листка райдужки, заднього ембріонтаксона і мезодермальних перемичок. Синдром Ригера – спадкове захворювання з домінантним типом передачі та високою експресивністю. Зустрічаються сімейні випадки.
Синдром Штурге-Вебера – енцефалотригемінальний ангіоматоз (гемангіоз шкіри, мозку та органу зору). Він характеризується капілярною гемангіомою обличчя вздовж розгалужень трійчастого нерва, включаючи повіки, орбіту, волосяну частину голови, наявні вогнищеві петрифікати в головному мозку. Від ступеня інтракраніальної патології залежить вірогідність неврологічних симптомів (епілепсія, гідроцефалія, парези, психічні порушення).
Глаукома розвивається тільки в тому випадку, коли уражені повіки, особливо верхні, і перебігає як доброякісний гідрофтальм з помірним збільшенням очного яблука.
При цьому синдромі ураження частіше буває одностороннім. В літературі приведені дані досліджень, що проводились проятогом 30 років, про наявність вродженої глаукоми в 72% випадків хворих синдромом Штурге-Вебера. В сучасній літературі наявні дані про вкрай несприятливий перебіг глаукоми при цьому синдромі.
Етіологія:
На даний час вивчена недостатньо. Етіологічними факторами можуть бути інфекційні, токсичні, аліментарні, нейросудинні, трофічні порушення, що впливають через материнський організм на плід, викликаючи різні ембріопатії з наступним розвитком типової вродженої глаукоми.
Деякі автори надають великого значення інфекційно-токсичним чинникам (сифіліс, токсоплазмоз, вірусні інфекції).
Розвиток глаукоми пов’язується з вірусними та паразитарними захворюваннями, перенесеними жінкою в перші місяці вагітності (краснуха, кір, поліомієліт, паротит, токсоплазмоз).
За останні роки приділяється велика увага гіпо- і авітамінозу, особливо дефіциту вітаміну А.
Не останнє місце займає ендокринна патологія – тиреотоксикоз і, безумовно, рефлекторна участь нервової системи. Підтвердженням ролі спадковості у хворих з вродженою глаукомою є наявність не тільки дефектів розвитку ока, але й всього організму.
Патогенез:
В основу патогенезу більшістю авторів покладена ембріологічна теорія, яка пояснює дитячу глаукому зупинкою зворотнього розвитку мезодерми, що заповнює у плода куток передньої камери.
Отже, основним патогенетичним фактором вродженої глаукоми є зміна мезенхіми з наявністю мезодермальної тканини в кутку передньої камери і патологічне диференціювання трабекули, дефект розвитку склеральної шпори, аномалії положення самого шлемового каналу, кореня райдужки та ціліарного м’яза слід вважати безпосередньою причиною порушення фільтрації вологи і приводом до підвищеного внутрішньоочного тиску.
Популяційна частота: невідома.
Співвідношення статей: Ч3 : Ж1.
Тип успадкування: аутосомно-рецесивний.
Диференційний діагноз: вторинна глаукома, паренхіматозний кератит, мегалокорнеа, ідіопатична едема.
Діагностика:
На пізніх стадіях розвитку не викликає сумнівів через характерні клінічні симптоми. Застосовується добова тонометрія (у маленьких дітей можлива тільки в стані наркотичного сну). Застосовується гоніоскопічний метод (виявлення мезодермальної тканини в кутку передньої камери). Показана ультразвукова діагностика.
Профілактика: медико-генетичне консультування.
Консервативне – малоефективне, до нормалізації тиску не призводить і замінити хірургічне лікування не може.
Хірургічне – ефективніше консервативного, але наявний ризик ускладнень (масивні кровотечі, атрезії очного яблука). Наявні методики не задовільняють офтальмохірургів результатами операцій. Найбільший відсоток успішних операцій зі стійким зниженням тиску та збереженням гостроти зору у віддалених після лікування випадках спостерігаються у дітей віком до 1 року.
Прогноз:
В результаті лікування в загальноосвітніх школах навчається половина хворих (частина з них за рахунок здорового ока при односторонньому видаленні), 32,4% пацієнтів навчаються в школах для дітей з порушенням зору, 17,6% – в школах для сліпих.
Номер з каталогу МІМ:
600975 Glaucoma 3, Primary Infantile, B; GLC3B.
Література:
- Кашинцева Л.Т., Кушине В.Л. Глаукома у больных с синдромом Стерджа-Вебера //Офтальмологический журнал.- 2002.- №4.
- Козлова С.И., Демикова Н.С., Семанова Е., БлинниковаО.Е. Наследственные синдромы и медико–генетическое консультирование.- М.: Практика, 1996.- C. 80-81.
- Сидоров Э.Г., Мирзаян М.Г. Врожденная глаукома и ее лечение.- М.: Медицина, 1991.
- Эрошевский Т.И., Токарева Б.А. Врожденная детская глаукома и ее лечение.- М.: Медицина, 1971.
Переглянуто редакційною колегією I.B.I.S.: 12/03/2003
Вплив кокаїну на плід
(Cocaine and the Fetus)
Сергій Лапченко
Інформаційний спеціаліст Волинського ОМНІ-центру
До теперішнього часу інформація про вроджені вади розвитку, спричинені наркотиками, хімічними речовинами та забрудненим навколишнім середовищем (так званими тератогенами), була неповна і іноді не зовсім вірна. Проте батьки, які бажають мати здорових і щасливих дітей, повинні знати і оцінювати небезпеку, яку ці речовини становлять для їх ще ненароджених дітей.
Серед усіх наркотиків найбільший вплив на плід має кокаїн. За останні кілька років дуже збільшилась кількість вагітних жінок, які вживають кокаїн і, відповідно, кількість новонароджених дітей, на яких діяв цей препарат. Багато з них народилися з серйозними медичними проблемами.
Як кокаїн може зашкодити ненародженій дитині?
Кокаїн може подіяти на вагітну жінку та її плід багатьма шляхами. На перших місяцях вагітності він може викликати викидень. Коли цей наркотик вживається в кінці вагітності, він може викликати передчасні роди, смерть плода або нанести непоправну шкоду головному мозку.
Дослідження доводять, що жінки, які під час вагітності вживали кокаїн, втричі частіше мали передчасні пологи і, як наслідок, недоношених дітей. Кокаїн зменшує надходження поживних речовин та кисню до дитини, отож, якщо навіть дитина доношена, вона все одно може народитися меншою ніж звичайно.
Кокаїн може також спричинити відшарування плаценти від стінки матки ще до того, як почнуться роди. Такий стан може призвести до сильної кровотечі і може бути смертельним як для матері, так і для дитини.
Як вживання кокаїну під час вагітності може вплинути на новонародженого?
Діти, які були під впливом кокаїну до свого народження, починають життя з серйозними медичними проблемами. Більшість з них народжуються дуже маленькими і занадто швидко. Вони часто мають низьку вагу при народженні (менше 2500 г). Такі діти помирають протягом першого місяця життя в 40 разів частіше ніж діти, народжені з нормальною вагою.
Деякі діти, на яких діяв кокаїн до народження, мають пошкодження мозку, неврологічні та дихальні проблеми. В деяких випадках їхні внутрішні органи можуть бути недорозвинені або аномальні.
Зразу ж після народження ці діти проходять через стан, подібний до “ломки” у наркоманів. Вони дуже нервові, боязкі та дратівливі, вони здригаються і кричать при найменшому доторкуванні чи від найменшого звука. Відповідно, таких дітей дуже тяжко заспокоїти, пізніше вони стають замкнутими та байдужими до всього. В результаті, вони часто не здатні на спілкування з оточуючими чи не реагують на своїх матерів. Така реакція дитини на кокаїн, подвоєна залежністю матері від наркотика, часто утруднює зв’язок між матір’ю і дитиною, що є важливим для емоційного розвитку дитини. Мати, в свою чергу, має проблеми з дитиною, яка весь час кричить, і її дуже тяжко заспокоїти. Таким чином, діти, народжені у жінок, які вживали наркотики, становлять групу ризику щодо дітей, які залишаються без батьківської опіки.
Які ще проблеми стоять перед дітьми, матері яких вживали кокаїн під час вагітності?
Ми не знаємо всіх проблем, з якими прийдеться зіткнутися таким дітям. Дослідження доводять, що вони мають набагато більший ризик померти від синдрому раптової смерті (СРС). В одному з таких досліджень стверджується, що таким чином померло 15% дітей, матері яких вживали кокаїн під час вагітності. В нормі цей показник становить лише 0,3%.
Спеціалісти прогнозують, що такі діти будуть мати підвищений ризик виникнення різних фізичних недоліків та нездатності до навчання в шкільному віці.
Переглянуто редакційною колегією I.B.I.S.: 15/02/2002
Синдром Вольфа-Хіршхорна
(Wolf-Hirschhorn Syndrome)
Т.Є. Зерова-Любимова
Старший науковий співробітник кафедри медичної генетики Київської медичної
академії післядипломної освіти ім. П.Л. Шупика Міністерства охорони здоров’я України
Включення:
Cиндром часткової делеції хромосоми 4.
Виключення:
Хромосома 5, моносомія 5р, хромосома 13, трисомія 13.
Основні діагностичні критерії:
Синдром Вольфа-Хіршхорна характеризується глибокою затримкою психомоторного та фізичного розвитку. Діти народжуються з низькою вагою. В перші місяці у них відмічається слабкий крик. Спостерігається мікроцефалія, асиметрія черепа, гіпертелоризм, опущені донизу зовнішні кути очних щілин, розщілина піднебіння, дзьобоподібний ніс з виступаючим надпереніссям, маленький рот з опущеними кутами (карпоподібний рот), деформовані вушні раковини з преаурикулярними виростами, гіпоспадія, затримка окостеніння зап’ясть та тазу.
При проведенні хромосомного аналізу делеція короткого плеча хромосоми 4 підтверджує діагноз.
Симптоми:
В більшості випадків спостерігається низька вага (2000 г) при народженні при доношеній або переношеній вагітності, глибока затримка психомоторного розвитку, затримка росту, мікроцефалія, гіпертелоризм, широкий або дзьобоподібний ніс, низько розташовані вушні раковини, мікрогнатія, гіпопластичні дермальні гребені, судоми, гіпоспадія у хлопчиків. Більше, ніж в половині випадків присутні такі риси, як птоз, епікант, антимонголоїдний розріз очей, колобома, страбізм, розщілина губи, піднебіння або язичка (увули), преаурикулярні складки, невеликі гемангіоми шкіри на обличчі – найчастіше на лобі, вади серця та ортопедичні аномалії – деформація ступнів.
Ускладнення:
Судоми, які важко контролювати і які можуть призвести до смерті пацієнта.
Поєднані аномалії:
Серед вад внутрішніх органів відмічають вроджені вади серця в 50% випадків, нирок, шлунково-кишкового тракту та дефекти центральної нервової системи (1/3 випадків).
Етіологія:
Даний синдром обумовлений делецією певного генетичного матеріалу короткого плеча хромосоми 4. Розрив хромосоми під час гаметогенезу призводить до реципрокної транслокації, а наступна сегрегація хромосом в мейозі дає гамету з делетованою хромосомою. Носій збалансованої транслокації може мати нащадків з такою ж транслокацією або з делецією (як при 4р- синдромі), або може зустрічатись і без транслокації, при цьому може делетуватись як кінець хромосоми, так і інтерстиціальний сегмент.
Виявилось, що при наявності делеції сегменту 4р16 відбувається повна експресія фенотипу даного синдрому. Для родин з реципрокними транслокаціями, в яких бере участь коротке плече хромосоми 4, існує високий ризик мати дітей з незбалансованими хромосомами.
Більшість випадків є спорадичними. Приблизно 13% описаних випадків обумовлені хромосомними абераціями у батьків, в основному транслокаціями.
Патогенез:
Відомо, що багато факторів, зокрема, хімічні та радіаційні, призводять до розриву хромосом. Водночас невідомі фактори обумовлюють розриви у людини in vivo. Вік матері та батька також не відіграє ролі.
Статеве співвідношення:
Частіше народжуються дівчата, ніж хлопці у співвідношенні 2:1.
Частота виникнення: 1 випадок на 50000.
Вік маніфестації:
Виявляються при народженні або в другому триместрі вагітності за допомогою аналізу культивування фібробластів плоду.
Життєвий прогноз:
Серед описаних випадків відомі мертвонародження та перинатальна смерть. Приблизно 35% помирають в перші два роки життя. Описані дорослі з даною патологією.
Генне картування – хромосомний аналіз – носійство:
Вперше синдром моносомії по короткому плечу хромосоми 4 описали Вольф та Хіршхорн із співавторами в 1965 році незалежно один від одного.
При цитогенетичному аналізі виявляють втрату частини матеріалу короткого плеча хромосоми 4, причому критичним сегментом для виникнення даного синдрому є сегмент 4р16 (Wilson et al., 1981), молекулярним аналізом в 1998 році було уточнено сегмент – 4р16.3 (Wright et al., 1998).
Довжина делетованої ділянки складає приблизно половину довжини короткого плеча хромосоми 4.
Лікування: невідоме.
Запобігання:
Носіїв збалансованих транслокацій можна виявити хромосомним аналізом.
Номер з каталогу МІМ:
194190 Wolf-Hirschhorn Syndrome; WHS.
602952 Wolf-Hirschhorn Syndrome Candidate 1; WHSC1.
Список літератури:
- Gandelman KY, Gibson L, Meyn MS, Yan-Feng TL. Molecular definition of the smallest region of deletion overlap in the Wolf – Hirschhorn syndrome. Am.J.Hum.Genet. 1992;51:571-578.
- Wilson MG, Towner JW, Ebbin AJ, Siris E, Berger P. Genetic and clinical studies in 13 patients with the Wolf – Hirschhorn syndrome del (4p). Hum. Genet. 1981;59:297-307.
- Wright TJ, Clemens M, Quarell O, Altherr MR. Wolf – Hirschhorn and Pitt – Rogers – Danks syndromes caused by overlapping 4p deletions. Am.J.Hum.Genet. 1998;75:345-350.
Переглянуто редакційною колегією I.B.I.S.: 20/12/2000
Вогневий невус
(Nevus Flammeus)
Т. Голуб
Лікар-неонатолог
Рівненської обласної дитячої лікарні
Включення:
Вогневий невус, nevus flammeus, “винні плями” або плями кольору “портвейну”, капілярні ангіодисплазії, телеангіектатичний невус.
Визначення:
Вогневий невус – це вада розвитку дерми, яка має вигляд червоної або фіолетової плями неправильної форми.
Основні діагностичні критерії, клінічні форми:
Невус спостерігається при народженні і ніколи не розсмоктується самостійно (за виключенням невуса Унни). Має вигляд плями рожевого, червоного або фіолетового кольорів неправильної форми. З віком у хворого можуть з’являтися папули або вузли. Великі невуси, зазвичай, обмежені одним або декількома сусідніми дерматомами і рідко пересікають середню лінію тіла. В 85% – однобічна локалізація. Може бути ураженою будь-яка ділянка тіла, але найчастіше – обличчя.
Невус Унни (вроджена телеангіектазія на потилиці) виявляють на задній поверхні шиї приблизно у 30-50% новонароджених. Інколи (10%) виникає на повіках над переніссям. Це єдина різновидність вогневого невуса, яка розсмоктується самостійно, переважно до 3-річного віку.
Синдром Штурге-Вебера. Поєднання вогневого невуса в зоні іннервації трійничного нерва з вадами розвитку судин ока (глаукома) та м’якої і павутинної мозкових оболонок (кальцинати, епілепсія, розумова відсталість, геміпарези).
Синдром Кліппеля-Треноне. Поєднання вогневого невуса з вадами розвитку судин м’яких тканин і кісток (вроджене варикозне розширення вен, гіпертрофія однієї або декількох кінцівок).
Синдром Кобба. Вогневий невус по задній серединній лінії у поєднанні з вадами розвитку судин спинного мозку (неврологічні розлади).
Гіпертрофія “винних плям” призводить до асиметрії обличчя або кінцівок, що вимагає ортопедичної корекції. Різниця у товщині та довжині кінцівок може становити декілька сантиметрів. Прогресуюча гіпертрофія “винних плям” може спричиняти закриття слухових каналів, носових ходів, звуження поля зору, тим самим порушуючи життєво важливі функції.
Діагноз:
Достатньо клінічної картини. Огляд обов’язково повинен включати консультацію невропатолога і вимірювання внутрішньоочного тиску ока.
Допоміжні дослідження:
Патоморфологія шкіри. Вада розвитку – розширення капілярів. Проліферації ендотеліальних клітин немає.
Променева діагностика. При синдромі Стерджа-Вебера рентгенографія черепа виявляє звапнені гемангіоми мозкових оболонок і лінійні вогнища звапнення по ходу звивин мозку. Необхідна КТ голови.
Диференційний діагноз: кавернозні гемангіоми.
Асоційовані вади:
Трисомії 13-15, синдром Тернера, вади розвитку судин очей, м’якої і павутинної мозкових оболонок.
Тип успадкування: аутосомно-домінантний.
Співвідношення статей: Ч1 : Ж1.
Частота: зустрічається у 0,3% новонароджених.
Вік прояву: вроджене захворювання.
Прогноз:
“Вогневий” невус самостійно ніколи не розсмоктується, крім невуса Унни. З віком у дітей розмір невуса збільшується. У дорослих поверхня невуса, зазвичай, стає горбкуватою і підвищується над рівнем шкіри, на ній з’являються папули та вузли, що спотворює хворого.
Можливі ускладнення:
Іноді може виникати базальноклітинний рак.
Лікування:
Доки не з’явилися папули та вузли, вогневий невус можна легко замаскувати за допомогою косметичних засобів. Ефективна лазерна терапія. В особливо важких випадках застосовують хірургічне втручання.
Номер з каталогу МІМ:
163100 Nevus Flammeus of Nape of Neck.
Література:
- Власов П.Г. Селективная коагуляция сосудистых дефектов лица лазерной установкой на парах меди “ЯХРОМА-М” //Ангиология и сосудистая хирургия.- 2001.- Том 7.- № 3.- C.100-104 (http://yachroma.com/exprnc31.htm).
- Козлова С.И., Семанова Е., Демикова Н.С. Наследственные синдромы и медико-генетическое консультирование: Справочник.– Л.: Медицина, 1987.- C.139.
- Куликов С.В., Поспелов Н.В., Пономарев О.Ю. Возможности лечения сосудистых патологий кожи лазером //Лечащий врач.- 2000.- № 5-6.- C.79-80 (https://www.lvrach.ru/2000/05-06/4526095/).
- Шабалов Н.Т. Неонатология. Учебник для студентов.- Издание второе.- Специальная литература, 1997. – C.464.
- Cloherty JP, Stark R. Manual of Neonatal Care. Fourth Edition. 1998:637-638.
Переглянуто редакційною колегією I.B.I.S.: 13/08/2004
Неімунна водянка плода та новонародженого
(Nonimmune Hydrops Fetalis)
І.Н. Мороз
Лікар-неонатолог
Відділення патології новонароджених Рівненської дитячої обласної лікарні
Включення:
Неімунний набряк.
Визначення:
Неімунний набряк – виражена загальна гідратація плоду, що проявляється позаклітинним скупченням рідини у тканинах і серозних порожнинах без будь-яких ознак циркулюючих антитіл до антигенів еритроцитів.
Основні діагностичні критерії:
Плевральний, перикардіальний випіт, асцит, периферійні набряки у плода, багатовіддя або потовщення плаценти (більше 6 мм). При виявленні більше, ніж однієї з вище вказаних ознак, використовують термін “водянка”.
Диференційний діагноз:
Диференційний діагноз проводиться з набряком плода та новонародженого імунного генезу (гемолітична хвороба плода і новонародженого).
Етіологія і патогенез:
Поява набряку обумовлена причинами, які призводять до змін в розподілі рідини між внутрішньо-судинними та інтерстиціальними позаклітинними просторами. У більшості випадків набряк виникає при зниженні білкового складу сироватки, що призводить до зниження тиску у капілярах.
Набряки можуть утворюватись при змінах у вмісті електролітів в поза-судинній і судинній рідині, що створює різницю в остмотичному тиску.
У недоношених новонароджених набряк спричиняється зниженням здатності виведення води та натрію, але в деяких випадках походження його невідоме.
У новонароджених набряк голови і шиї може бути наслідком обвиття шиї пуповиною, а транзиторні обмежені набряки кистей та ступнів виникають внаслідок внутрішньоутробного стиснення. Неімунна водянка може супроводжувати серцеву недостатність, пов’язану з вродженими захворюваннями серця, навіть при відсутності шумів.
Майже в 44% причину виникнення неімунної водянки встановити не вдається і її вважають ідіопатичним станом.
Частота:
1 : 1500-4000 новонароджених. В даний час, в зв’язку з проведеннями профілактичної Rh-ізоімунізації жінок, неімунна водянка зустрічається в 6-7 разів частіше, ніж імунна водянка плода.
Асоційовані аномалії:
Патологічні стани під час вагітності, що супроводжуються розвитком набряку плоду, поділяються на 4 групи:
- Захворювання плоду.
- Патологія серцево-судинної системи:
- Порушення серцевого ритму (суправентрикулярна тахікардія, фібриляція передсердь, атріовентрикулярна блокада, складні аритмії);
- Захворювання міокарда (ендокардіальний фіброеластоз, кардіоміопатія, інфаркт при тромбозі коронарних артерій серця, єдиний шлуночок серця, загальний передсердно-шлуночковий канал, внутрішньоутробний міокардит);
- Вади розвитку серця (дефекти міжшлуночкової та міжпередсердної перетинок, тетрада Фалло, гіпоплазія лівих відділів, стенози і атрезії клапанів);
- Патологічні утвори в порожнинах серця (пухлини – рабдоміома, міксома; тромби);
- Передчасне закриття судинних шунтів (артеріальна протока, овальний отвір).
- Патологія, що спричиняє розвиток анемії у плода (хронічні плодово-материнські кровотечі, хронічна плодово-материнська трансфузія при монохоріальній багатоплідній вагітності, гомозиготна альфа-таласемія, дефіцит глюкозо-6-фосфатдегідрогенази, травми плоду, кровотечі при інвазивних втручаннях).
- Позасерцева патологія розвитку органів грудної клітки:
- Легені (кістозно-аденоматозна вада розвитку легень, лейоміосаркома, аденома, позадольова секвестрація легень, гіпоплазія легень);
- Патологія лімфатичної системи (лімфангіоектазія, розрив лімфатичних судин, хілоторакс);
- Діафрагмальна кила.
- Патологія шлунково-кишкового тракту і органів черевної порожнини:
- Аномалії розвитку, патологія печінки та її судинної системи (фіброз, вроджений гепатит, цироз, кальциноз, портальна гіпертензія, гемангіоендотеліома);
- Аномалії розвитку і патологія кишки (атрезії будь-яких відділів кишки, меконіальний перитоніт, дивертикул дванадцятипалої кишки, подвоєння кишки);
- Аномалії розвитку селезінки (аспленія, поліспленія).
- Патологія сечовивідної системи плоду:
- Аномалії розвитку нирок (гіпоплазія, дисплазія, вроджений нефротичний синдром, полікистоз, тромбоз вен);
- Порушення прохідності сечовивідних шляхів (стеноз чи аплазія уретри, задньоуретральний клапан, гідронефроз, самовільний розрив сечового міхура, перфорація сечового міхура при шунтуванні);
- Аномалії розвитку статевого апарату (атрезія піхви, гіпоплазія матки, подвоєння матки, сечостатевий синус);
- Кіста яйника.
- Неоплазії різної локалізації (нейробластома, тератоми (крижова, медіастінальна), туберозний склероз).
- Внутрішньоутробна інфекція (TORCH – токсоплазмоз, краснуха, цитомегаловірус, простий герпес, сифіліс, лептоспіроз, парвовірус В-19).
- Патологія каріотипу і генні мутації (трисомії 13, 18, 21, триплодія (69ХХХ), синдром Тернера (45 ХО), мозаїцизм (46XX/XY), хромосомні транслокації, міодистрофія Дюшена-Беккера).
- Патологія ЦНС (гідроцефалія, внутрішньошлуночкові крововиливи, енцефалоцеле, поренцефалія з агенезією мозолистого тіла).
- Метаболічні порушення (хвороба Тей-Сакса, хвороба Гоше, GM1-гангліозидоз, мукополісахаридоз).
- Скелетні дисплазії (ахондроплазія, ахондрогенез, незавершений остеогенез, асфіктична дисплазія грудної клітини, артрогрипоз, синдром Нунан, карликовість).
- Багатоплідна вагітність (паразитуючий плід, акардія-ацефалія плода).
- Інші.
- Патологія серцево-судинної системи:
- Патологія плаценти і пуповини.
- Пухлини плаценти (хоріоангіома).
- Пухлини пуповини (ангіоміксома).
- Порушення розвитку чи прохідності судин плаценти і пуповини (тромбоз вен хоріону, оклюзія артерії пуповини, аневризма пуповини, артеріо-венозні шунти судин плаценти).
- Захворювання матері.
- Важкі анемії різної етіології.
- Важка гіпертензія, токсикози.
- Цукровий діабет.
- Ідіопатичні форми. У новонароджених відмічається набутий генералізований синдром набряків при таких станах:
- Надлишкова чи неадекватна інфузійна терапія;
- Гіпопротеїнемія;
- Синдром неадекватної підвищеної секреції антидіуретичного гормону;
- Ятрогенний нейром’язевий параліч;
- Гіпотиреоз;
- Надлишок стероїдних гормонів;
- Обмінні порушення – грубі дефіцити цинку, вітамінів В-групи, РР та інших;
- Серцева недостатність, обумовлена як захворюваннями серця (вроджені вади, міокардити тощо), так і електролітними порушеннями, важкою гіпоксією, легеневою гіпертензією, поліцитемією, синдромом гіперв’язкості;
- Гостра ниркова недостатність (вади розвитку або тромбоз ниркових судин);
- Тромбози магістральних судин.
Прогноз:
Незважаючи на те, що в літературі повідомляється про деякі випадки спонтанної внутрішньоутробної ремісії водянки плода, прогноз у більшості випадків буває несприятливий. Рівень перинатальної смертності при цьому дуже високий і складає 70-90%.
Акушерська тактика при діагностуванні неімунної водянки плода:
В разі виявлення неімунного набряку для визначення тактики ведення вагітної рекомендоване комплексне обстеження стану плоду та вагітної жінки. (див. cхему 1).
Схема 1. Алгоритм обстеження при виявленні набряку плоду
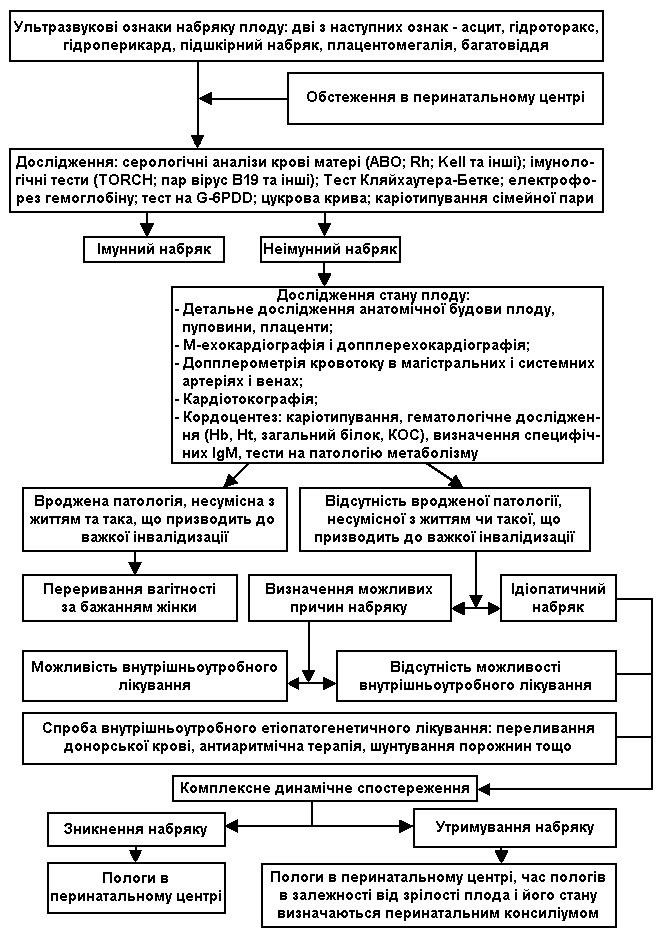
Враховуючи високу частоту випадків хромосомної патології (30-40%), у всіх випадках показано пренатальне каріотипування. Виявлення вродженої патології, несумісної з життям або такої, що призведе до важкої інвалідизації, є показом для переривання вагітності. При відсутності вказівок на наявність такої патології показане динамічне спостереження за станом плоду, враховуючи можливість самовільного зникнення набряку, навіть при відсутності внутрішньоутробної терапії. У випадку виявлення патогенетичних механізмів розвитку неімунної водянки при наявності потенційної можливості їх корекції, проводять внутрішньоутробну консервативну медикаментозну чи інвазивну терапію. У випадках самовільного зникнення набряку у плода при динамічному спостереженні родорозрішення проводять у відповідний термін в умовах перинатального центру. При збереженні набряку рішення про ведення пологів приймається перинатальним консиліумом після оцінки стану плода, зрілості його органів і систем. Якщо вагітність закінчується в строк, вибір методу ведення пологів може знизити ризик внутрішньоутробної смерті плода. Асцит у плода може бути причиною дискоординованої пологової діяльності, тому методом вибору служить операція кесарського розтину, що забезпечить мінімальну пологову травму і максимальне виживання.
Номер з каталогу МІМ:
236750 Hydrops Fetalis, Nonimmune; NIHF.
Література:
- Клиническое руководство по ультразвуковой диагностике /Под ред. В.В. Митькова.- М.: Видар, 1996.- С. 174-177.
- Пренатальная диагностика врожденных пороков развития /Р. Ромеро, Дж. Пилу, Ф. Дженти, Дж.С. Хоббинс.- М.: Медицина, 1994.- С. 414-424.
- Руководство по педиатрии (болезни плода и новорожденного, врожденные нарушения обмена веществ) /Под ред. Р.Е. Бергмана, В.К. Вогана.- М.: Медицина,1987.- С. 260-261.
- Шабалов М.П. Неонатология.- Спб: Специальная литература, 1995.- С. 318-319.
Переглянуто редакційною колегією I.B.I.S.: 26/03/2002
Затримка внутрішньоутробного розвитку у дітей
(Intrauterine Growth Retardation)
Л.М. Грицута
Лікар-педіатр
Міська дитяча лікарня, м. Рівне
Включення:
Cиндром затримки розвитку плоду (СЗРП).
Визначення:
Затримку внутрішньоутробного росту та розвитку (ЗВУР) діагностують у дітей, які мають недостатню масу тіла при народженні по відношенню до їхнього гестаційного віку, тобто коли величина маси тіла нижча 10% центиля в терміні вагітності матері, або морфологічний індекс зрілості відстає на 2 чи більше тижні від справжнього гестаційного віку.
Доцільно оцінювати гестаційний вік дитини за сукупністю морфологічних ознак: станом шкіри, грудних залоз, геніталій, вух, ступнів, лануго (від 0 до 5 балів); а також за оцінкою нейром’язової зрілості. При цьому враховується положення немовляти; квадратне вікно (зап’ястя), тобто кут між внутрішньою поверхнею передпліччя і підвищенням великого пальця; зворотну реакцію руки дитини на згинання в ліктьовому суглобі; підколінний кут; симптом “шарфа”; протягування п’ятки до вуха (від 0 до 5 балів по Дж. Балларду і співавторам).
Згідно рекомендаціям ВООЗ, з 1961 року діти, які народилися з масою тіла менше 2500 грам, характеризуються як “мала вага при народженні”. Серед цих дітей виділяють 3 групи:
- Недоношені з масою тіла, яка відповідає їх гестаційному віку.
- Недоношені з масою тіла меншою, ніж за терміном вагітності.
- Доношені (народжені після 37 тижнів вагітності) або переношені (народжені після 42 тижнів вагітності), які мають малу масу тіла (нижче 10%) для даного терміну гестаційного віку.
Діти 1-ї групи, звичайно, досягають нормального внутрішньоутробного росту та розвитку, а діти 2 і 3 груп розвиваються із затримкою. Пренатальна гіпотрофія, тобто знижене харчування, виснаження, яке виникає внутрішньоутробно, є не у кожної дитини із ЗВУР (у 2 із 3). Пренатальна смертність дітей із ЗВУР в 3-5 разів більша, ніж у здорових дітей.
Етіологія:
4 групи факторів ризику, які призводять до СЗРП.
- Материнські – дефект харчування, хронічні хвороби, шкідливі звички, вживання медикаментів, обтяжений акушерський анамнез.
- Плацентарні – вади розвитку плаценти, аномалії, відшарування, недостатня маса і поверхня.
- Плодові – спадкові фактори, багатоплідна вагітність, вроджені вади розвитку, генералізовані внутрішньоутробні інфекції.
- Соціально-біологічні.
У 40% дітей виявити причину СЗРП не вдається. У США головною причиною ЗВУР є матково-плацентарна недостатність; 10% зафіксованих випадків обумовлені внутрішньоутробною інфекцією. У 5-15% новонароджених СЗРП – результат хромосомних захворювань та інших генетичних порушень.
Частота поширення СЗРП серед новонароджених складає, за даними різних авторів від 2 до 17,6%. 20% мертвонароджених мають СЗРП. Останнім часом відзначається тенденція до його збільшення, що пов’язано як з дійсним почастішанням цієї патології, так і з поліпшенням діагностики. За даними перинатальної статистики, в Україні частота СЗРП у різних регіонах складає від 10 до 22% серед доношених новонароджених та від 18 до 22% серед недоношених. В останні роки всебічному вивченню цієї патології приділяється велика увага, як в нашій країні, так і за кордоном. Це пов’язано з тим, що перинатальна захворюваність і смертність в значній мірі визначається характером росту й станом плода в гестаційному періоді і досягає 30%.
Класифікація:
У діагнозі СЗРП виділяють:
- етіологічні фактори і стани ризику (материнські, плацентарні, плодові, змішані);
- клінічний варіант (гіпотрофічний, гіпопластичний, диспластичний);
- ступінь важкості (легка, середня, важка);
- перебіг інтранатального і неонатального періодів (без ускладнень чи з ускладненнями).
Виділяють також:
- симетричний СЗРП (ОГ=Д=МТ, все менше 10%); oбвід голови (ОГ), довжина (Д) і маса тіла (МТ) пропорційно знижені по відношенню до нормальних показників для даного гестаційного віку; симетричний СЗРП обумовлений або зниженням потенційних можливостей плоду у рості (генетичне порушення, внутрішньоутробна інфекція), або ж екзогенними факторами, які діють у ранні терміни вагітності;
- асиметричний СЗРП (ОГ=Д>МТ, все менше 10%); маса тіла плоду зменшена непропорційно по відношенню до довжини та обводу голови; обвід голови і довжина ближчі до очікуваних перцентелів для даного гестаційного віку, ніж маса тіла; найпоширенішими причинами є матково-плацентарна недостатність, неповноцінне харчування матері чи екзогенні фактори, які діють у останні тижні внутрішньоутробного розвитку.
Клініка:
Гіпотрофічний варіант СЗРП. Пренатальна гіпотрофія. Клінічна картина залежить від ступеня важкості і включає в себе наступні ознаки: дефіцит маси тіла по відношенню до довжини; трофічні розлади шкіри; маса м’язів; об’єм голови; перебіг раннього неонатального періоду. Діти даної групи схильні до більшої втрати початкової ваги тіла і тривалого її відновлення, затяжної транзитної жовтяниці новонароджених, повільного заживання пупкової рани. У ранньому неонатальному періоді характерні порушення функції ЦНС.
Гіпопластичний варіант. Діти мають відносно пропорційне зменшення всіх параметрів фізичного розвитку. Виглядають пропорційними, але маленькими. Можуть бути поодинокі стигми. Шви голівки не закриті, краї тім’ячка м’які, піддатливі. У ранньому неонатальному періоді діти схильні до переохолодження, респіраторних розладів. Ступінь важкості гіпопластичного варіанту СЗРП визначають за дефіцитом довжини тіла і обводу голови по відношенню до терміну гестації: легкий – дефіцит 1,5-2 сигми, середньої важкості – більше 2, але менше 3 сигм, і важкий – більше 3 сигм.
Диспластичний варіант. Найчастіше – це прояв спадкової патології (хромосомні або генні аномалії), генералізованих внутрішньоутробних інфекцій, тератогенного впливу. Типовими проявами СЗРП цього варіанту є вади розвитку, порушення будови, стигми. У клініці характерні важкі неврологічні розлади, обмінні порушення, анемія, ознаки інфекції. Сюди відносяться синдром Дауна, синдром Едвардса, синдром Патау, синдром Шерешевського-Тернера та ін.
Діагностика:
Гестаційний вік визначають за сумою балів, які відображають стан морфологічної зрілості. Для виявлення гіпотрофії вираховують масо-ростовий індекс (Пондерала). Обстеження дітей зі СЗРП в неонатальному періоді направлене на виявлення:
- захворювань, симптомом яких може бути СЗРП;
- найтиповіших ускладнень.
У зв’язку з цим необхідні: клінічний аналіз крові, визначення КОС, гематокритного числа, глікемії, білірубіну та його фракцій, рівня загального білка і білкових фракцій, сечовини, калію, натрію, кальцію, магнію у сироватці крові; клінічний аналіз сечі; скринінг на найпоширеніші спадкові аномалії обміну речовин; ехоенцефалографія. При підозрі на внутрішньоутробне інфікування призначають відповідні дослідження.
Патофізіологія:
Внутрішньоутробний розвиток плоду визначається фетальними, материнськими і плацентарними факторами.
- Фетальні фактори СЗРП.
- Генетичні фактори:
- расові, етнічні та національні відмінності;
- генні захворювання;
- хромосомні хвороби;
- жіноча стать;
- вроджені вади розвитку.
- Внутрішньоутробні інфекції.
- Вроджені порушення метаболізму.
- Генетичні фактори:
- Материнські фактори СЗРП.
-
- Гіпертонія, індукована вагітністю (більше 140/90 мм рт. ст.).
- Набування ваги менше 0,9 кг кожних 4 тижня.
- Відставання у збільшенні висоти стояння дна матки (менше 4 см для даного гестаційного віку).
- Вада серця синього типу.
- Паління.
- Проживання на великій висоті над рівнем моря.
- Наркоманія та токсикоманія.
- Малий зріст.
- Низький соціально-економічний рівень.
- Анемія (гематокрит менше 30%).
- Маса тіла до вагітності менше 50 кг.
- Народження дітей з СЗРП від попередніх вагітностей.
- Хронічна гіпертонія.
- Хвороби нирок.
- Важке недоїдання.
- Багатоплідна вагітність.
- Молодий вік матері.
- Знижений матково-плацентарний кровообіг. Такі захворювання матері, як прееклампсія-еклампсія, хронічна гіпертонія і хронічний гломерулонефрит часто призводять до зниження матково – плацентарного кровообігу і пов’язаного з цим СЗРП. Недостатня кількість кисню і необхідних поживних речовин обмежує ріст внутрішніх органів і дозрівання скелету і мускулатури.
- Недостатнє харчування матері. Недостатнє харчування матері призводить до дефіциту поживних речовин, які поступають до плоду. Загальна енергетична цінність поживних речовин більше впливає на масу дитини при народженні, ніж на споживання білків та жирів. Голод призводить до помірного зменшення маси тіла при народженні; недостатнє харчування матері є найчастішою причиною СЗРП у країнах третього світу. Найбільш несприятливий вплив на масу тіла дитини при народженні чинить голодування матері в останньому триместрі вагітності.
- Багатоплідна вагітність. Затримка росту плоду може бути результатом неможливості забезпечити оптимальне харчування більше, ніж одного плоду. При цьому відмічається прогресивне зниження маси тіла дітей з двійні чи трійні. При парабіотичній двійні близнюк-донор отримує недостатню кількість поживних речовин внаслідок змін плацентарного кровообігу, обумовлених наявністю артеріовенозних шунтів в плаценті.
- Наркотики та препарати.
- Цигарки та алкоголь. Безумовним є зв’язок між СЗРП та вживанням цигарок та алкоголю. Негативні ефекти алкоголю та тютюну залежать від вживаної кількості: ступінь СЗРП вищий і вірогідність розвитку вища при використанні великих доз алкоголю та нікотину.
- Героїн. Вживання матір’ю героїну також часто супроводжується розвитком СЗРП у плоду.
- Інші. До інших наркотиків та хімічних препаратів, що викликають СЗРП, відносяться такі відомі тератогенні речовини, як антиметаболіти, та такі терапевтичні препарати, як триметадіон, варфарин, фенітоін. Кожний з цих агентів приводить до формування специфічних вад розвитку.
- Гіпоксемія у матері. У районах, розташованих високо над рівнем моря, виражена тенденція до народження дітей, середня маса тіла яких нижча, ніж у дітей цього ж гестаційного віку, що народились на рівні моря.
- Інші материнські фактори. Малий зріст матері, низька вага до вагітності, молодий вік, низький соціально – економічний рівень життя, перша вагітність, численні вагітності в анамнезі є причинами народження дітей, які мають масу тіла нижчу за норму.
-
- Плацентарні фактори.
- Плацентарна недостатність. У першому та другому триместрах вагітності ріст плоду визначається головним чином спадковою детермінованістю. У третьому триместрі головне значення у визначенні росту плоду починають займати плацентарні фактори, тобто фактори, що забезпечують плід адекватною кількістю поживних речовин. Коли тривалість вагітності починає перевищувати можливості плаценти забезпечувати плід поживними речовинами, розвивається плацентарна недостатність з наступною затримкою росту плоду. Цей феномен зустрічається переважно при переношеній вагітності, але може розвинутись і в будь – якому терміні вагітності.
- Анатомічні проблеми. Різноманітні анатомічні дефекти, такі як численні інфаркти, аномальне випадання пуповини, тромбоз пупкових судин і гемангіоми, описані при морфологічному обстежені плаценти у випадках СЗРП. Передчасне відшарування плаценти призводить до зменшення площі поверхні, яка бере участь в обміні речовин між матір’ю та плодом, обумовлюючи затримку його розвитку. Порушення умов внутрішньоутробного розвитку відображається як на рості плоду, так і на рості плаценти, тому новонароджені з СЗРП зазвичай мають маленьку плаценту.
- Плацентарні фактори СЗРП.
- Відшарування плаценти.
- Гемангіома.
- Єдина пупкова артерія.
- Інфаркт.
- Аномальне випадання пуповини.
- Тромбоз пупкових судин.
Пренатальна діагностика:
На даний час ультразвукова діагностика відкриває широкі можливості для виявлення СЗРП. Наступні параметри, які оцінюються комплексно, дозволяють з високим ступенем точності судити про затримку розвитку плоду:
- Біпарієтальний діаметр (БПД). Якщо при повторних вимірах БПД результат нижчий за оптимальний, вірогідність народження дитини з низькою масою тіла (по відношенню до очікуваної для даного гестаційного віку) складає 50 – 80%.
- Обвід живота. Печінка – орган, який страждає першим при затримці росту. Зменшення обводу живота – ранній симптом асиметричної затримки розвитку плода і знижених запасів глікогену.
- Відношення обводу голови до обводу живота. В нормі воно змінюється зі збільшенням терміну вагітності. В другому триместрі обвід голови більший, ніж обвід живота. Приблизно в 32-36 тижнів вагітності співвідношення складає 1:1, а після 36 тижнів обвід живота стає більшим. Збереження співвідношення більше 1 в останні тижні вагітності свідчить про наявність асиметричного СЗРП.
- Довжина стегна. Довжина стегна добре корелює з довжиною тіла, виміряної від маківки до п’ят, що дає можливість оцінювати довжину плоду на ранніх термінах і здійснювати потрібні вимірювання. Повторна оцінка довжини стегна, як і БПД, ефективна при встановлені симетричного СЗРП.
- Оцінка морфології плаценти і амніотичної рідини. Вона допомагає відрізнити плід з затримкою розвитку від плоду з конституційно обумовленими невеликими розмірами. Наприклад, ознаки старіння плаценти у поєднанні з маловіддям свідчить про СЗРП і про поганий прогноз для плода, в той час як нормальна морфологія плаценти і нормальна кількість навколоплідних вод передбачають конституційно обумовлені невеликі розміри плоду.
На схемі №1 представлено підходи до диспансерного спостереження за вагітними з СЗРП.
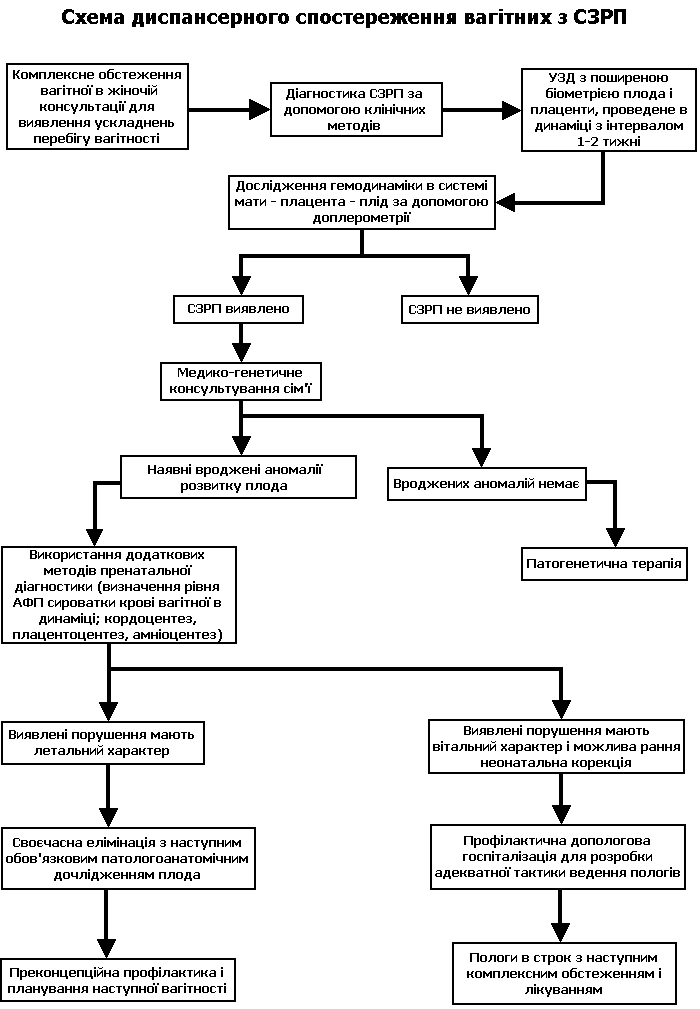
Лікування:
Враховуючи схильність до швидкого охолодження, дітей з СЗРП необхідно одразу ж після народження і в подальшому оглядати під променевим джерелом тепла. Далі їх розміщують у кювезі чи в грілці-ліжечку або ж зігрівають іншим способом (грілка до ніг, лампа солюкс). Об’єм і характер лікарської допомоги при народженні визначаються наявністю чи відсутністю первинної асфіксії.
Вигодовування. При наявності рефлексу смоктання, відсутності асфіксії, виражених неврологічних змін і зригування після пробного пиття дистильованої води дитину починають годувати зцідженим грудним молоком з пляшечки не пізніше, ніж через 2 години після народження. Кількість молока у різні дні життя визначаються за такими ж принципами, що і у недоношених дітей. При неможливості почати ентеральне годування необхідна інфузійна терапія. Термін прикладання до грудей залежить від маси тіла дитини при народженні, її стану, супутніх захворювань та ускладнень.
Медикаментозна терапія. Всім дітям з СЗРП одразу ж після народження парентерально вводять 1-2 мг вітаміну К. Важливо з першого дня призначити для внутрішнього прийому біфідумбактерин по 1 дозі 2 рази в день. При важких ступенях СЗРП, народженні дитини в асфіксії, перших ознаках СДР корисно з першого дня життя (найкраще в перші години життя) щоденно внутрішньом’язово протягом 3-5 днів вводити вітамін Е (по 20 мг/кг). Характер і об’єм терапії залежать від перебігу пологів, наявності ускладнень, супутніх захворювань. При недостатньому набиранні маси тіла необхідно зробити копрограму і за її результатами вирішити, чи необхідна замісна терапія ферментами (абомін, фестал, панкреатин та ін.).
Лікування плоду з СЗРП починається з визначення причини. Якщо у жінки виявлені дефіцит харчування, гіповітаміноз, то їх потрібно ліквідувати або корегувати. При матково-плацентарній недостатності корисні абдомінальна декомпресія, киснева терапія (нерідко при цьому відмічають швидке покращання росту плоду), курс пірацетаму.
Прогноз:
Залежить від варіанту і ступеня важкості СЗРП. При гіпотрофічному та гіпопластичному варіантах І ступеня діти, що не мали важких ускладнень в анте- і неонатальному періодах, як правило, доганяють своїх ровесників (які відповідають гестаційному віку при народженні і не мали СЗРП) у фізичному розвитку до 6 місяців життя, рідше – у другому півріччі. Вони можуть відставати у психомоторному розвитку. Інфекційна захворюваність не перевищує захворюваність ровесників без СЗРП. При ІІ ступені СЗРП більшість дітей доганяють своїх ровесників за фізичним розвитком до 1 року, в деяких з них підвищена інфекційна захворюваність і відставання в психомоторному розвитку у перші 2 роки життя. Рано розвиваються залізодефіцитна анемія, рахіт, а в подальшому спостерігаються ознаки легкої мозкової дизфункції (невропатичні порушення, інфантилізм психіки, невротичні реакції і ін.). У дітей з ІІІ ступенем СЗРП, а також з його диспластичним варіантом прогноз завжди потрібно робити з обережністю. Відставання фізичного та психомоторного розвитку в них може бути тривалим (до 3-4 років і більше), у 10-15% з них в подальшому розвиваються ознаки органічного ураження ЦНС (дитячий церебральний параліч, епілепсія, прогресуюча гідроцефалія, відставання в психічному розвитку і навіть олігофренія та ін.). При ІІІ ступені СЗРП дуже висока інфекційна захворюваність вже в неонатальному періоді, кожна 3-4 дитина переносить сепсис. У кінці 80-х – на початку 90-х років з’явились праці, в яких було доведено зв’язок СЗРП з розвитком в дорослому віці гіпертонічної хвороби, цукрового діабету. Генез цього зв’язку не встановлений, але гіпертонічну хворобу пов’язують з посиленим синтезом в перинатальному періоді ангіотензину ІІ (ангіотрофічний фактор для гладкої мускулатури судин, фактор росту судин під час внутрішньоутробного періоду) при хронічній внутрішньоутробній гіпоксії.
Література:
- Неонатология /Под ред. Т.Л. Гомеллы, Н.Д. Каннигам. – М.: Медицина, 1998.- С. 409-418.
- Шабалов Н.П. Неонатология. – СПб: Специальная литература, 1997.- Т.1.- С. 65-81.
- Яковенко О.Я. Пренатальна діагностика генетично обумовлених форм синдрому затримки розвитку плоду. Business information.- 1998.- № 33.- С. 44-50.
Переглянуто редакційною колегією I.B.I.S.: 06/02/2002
Синдром Вільямса – обвід голови у дівчат
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”.
Дані про обвід голови дівчаток з синдромом Вільямса, зібрані в результаті обстеження 61 пацієнтки. Пунктирними лініями на діаграмі показані нормальні темпи збільшення обводу голови; суцільною лінією – динаміка розвитку осіб з синдромом Вільямса. Діаграми створено Collen A.Morris, M.D., Susan A.Demsey, M.S., Clair O.Leonard, M.D., Constance Dilts, M.A., and Brent Blackburn, B.A. The Williams Syndrome National Association is gratefully acknowledged for their cooperation.
|
|
|
|
|
|






{kind=link}