
Uncategorized
Спинно-мозкова кила
(Spina Bifida)
Сергій Лапченко
Спеціаліст з інформаційного забезпечення
ОМНІ-Мережі для дітей
Спинно-мозкова кила, або, як її часто називають, відкритий хребет, – це вроджена вада розвитку, при якій хребет та спинно-мозковий канал не закриваються перед народженням. Результатом цього є те, що спинний мозок та покриваючі його тканини виступають за межі спини дитини.
Як спинно-мозкова кила впливає на дитину?
В нормі спинний мозок закривається на 29-й день вагітності. У випадку виникнення спинно-мозкової кили хребет повністю не закривається. Є три форми цієї вади розвитку.
- Скрита форма (occulta). При цій звичайно бессимптомній формі спинно-мозкової кили є тільки невеликий дефект або отвір в одній чи кількох маленьких кістках хребта. Спинний мозок та нерви нормальні і більша частина хворих на цю форму спинно-мозкової кили не мають ніяких проблем, спричинених цією аномалією.
- Менінгоцеле. При цій формі спинно-мозкової кили утворюється киста (випираюча припухлість), яка складається з оточуючих спинний мозок оболонок. Ця киста виступає над відкритою частиною хребта. Спинний мозок та нерви звичайно знаходяться в нормальному стані. Розмір кисти може бути різним: від маленького горішка до великого яблука. В більшості випадків ця вада може бути виправлена за допомогою хірургічного втручання, надаючи можливість дитині розвиватися цілком нормально.
- Мієломенінгоцеле. При цій найбільш вираженій формі спинно-мозкової кили виступаюча частина складається з нервових закінчень спинного мозку, а часто навіть і з самого спинного мозку, покритих тонкою оболонкою. Іноді киловий мішок може бути відсутній, в цьому випадку спинний мозок і нерви будуть повністю відкритими. Може витікати спинно-мозкова рідина і в результаті виникнути запалення всіх оточуючих тканин. Такі діти мають високий ризик бути інфікованими, тому необхідне термінове хірургічне втручання для закриття отвору в спині. Також проводиться антибіотикотерапія з метою попередження інфекції. Незважаючи на лікування, такі діти можуть мати різні ступені паралічу ніг, сечового міхура, кишківника.
Що таке гідроцефалія?
Гідроцефалія, або збільшена кількість рідини в мозку, зустрічається у 70-90% дітей з найбільш вираженою формою спинно-мозкової кили. Це є результатом того, що спинно-мозкова рідина, яка в нормі циркулює і захищає головний та спинний мозок, не може нормально циркулювати через наявність кили. Таким чином, рідина збирається в мозку та навколо нього, викликаючи збільшення розмірів голови. Без лікування цей процес може закінчитися затримкою розумового розвитку та іншими неврологічними порушеннями. (Гідроцефалія у дітей буває також викликана іншими, не пов’язаними з спинно-мозковою килою, причинами).
Що викликає появу спинно-мозкової кили?
Як правило, спинно-мозкова кила не пов’язана з іншими вродженими вадами розвитку. Причини її виникнення до кінця ще не вияснені. Вважається, що вона є результатом як впливу генетичних факторів, так і навколишнього середовища. 95% дітей із спинно-мозковою килою та іншими вадами нервової трубки походять з сімей, які ніколи не мали в своєму родоводі подібних аномалій. Є сім’ї, які мають схильність дo появи у їхніх дітей спинно-мозкової кили, але поки що не виявлено якогось певного закону успадкування. Якщо батьки вже мають одну дитину із спинно-мозковою килою, ризик народження ще однієї дитини з цією вадою буде 2%. Якщо ж у батьків народилося двоє дітей із спинно-мозковою килою, то ризик зростає до 5%.
Чи можливо попередити виникнення спинно-мозкової кили?
Дослідження доводять, що на розвиток спинно-мозкової кили та інших вад нервової трубки впливає харчування матері, зокрема, кількість вітаміну В (фолієвої кислоти) у їжі. Лікарі рекомендують усім жінкам дітородного віку (від 15 до 44 років) приймати по 0,4 міліграма фолієвої кислоти в день протягом місяця до зачаття та перших 3-х місяців вагітності. Це зменшує ризик народження дітей із спинно-мозковою килою на 50-70%.
Оскільки більша частина вагітностей незаплановані (випадкові) і багато жінок протягом кількох тижнів не знають, що вони вагітні, необхідний щоденний прийом фолієвої кислоти.
Їжа не може забезпечити необхідну кількість фолієвої кислоти, але остання входить до складу багатьох полівітамінів. Треба тільки бути обережним, щоб не прийняти занадто багато полівітамінів. Приймайте стільки, скільки приписав Вам лікар.
Як лікується спинно-мозкова кила?
Легка форма спинно-мозкової кили (occulta) не потребує лікування. Менінгоцеле, при якому спинний мозок не страждає, може бути виправлене хірургічно. Гідроцефалія і проблеми з сечовим міхуром не характерні для менінгоцеле, хоча лікарям треба мати на увазі можливість їх виникнення і необхідність швидкого лікування. Більша частина дітей з менінгоцеле розвиваються нормально.
Якщо народжується дитина з тяжкою формою спинно-мозкової кили, її оперують одразу ж (протягом 24-48 годин після народження). Хірург звільняє спинний мозок, поміщає його назад в спинно-мозковий канал і закриває його м’язами та шкірою. Така швидка операція допомагає запобігти більшому ушкодженню нервів інфекцією або травмою. Однак, навіть швидке оперативне втручання не може відновити вже пошкоджені нерви і, в цьому випадку, може залишитися параліч кінцівок, сечового міхура та кишківника.
Одразу ж після операції лікар-фізіотерапевт покаже батькам, які вправи треба робити для ніг і ступнів дитини, щоб підготувати їх для ходіння з протезами. Майже 72% таких дітей можуть пересуватися самостійно за допомогою різних засобів.
Якщо у дитини виникла гідроцефалія, зайва рідина може бути видалена з головного мозку за допомогою спеціальної трубки, яка називається шунтом.
Після такого лікування діти зі спинно-мозковою килою звичайно стають активними членами суспільства. Більша частина жінок з цією аномалією можуть мати дітей, хоча такі вагітності вважаються вагітностями високого ризику.
Чи можна діагностувати спинно-мозкову килу до народження дитини?
Спинно-мозкова кила може бути діагностована до народження за допомогою 2 або більше тестів (обстежень).
- Більшість медиків пропонують зробити тестування крові за допомогою альфа-фетопротеїну (АФП). Цей тест вказує на імовірність виникнення спинно-мозкової кили або інших вад нервової трубки у плода. Якщо тест вказав на високий ризик виникнення патології, а термін вагітності визначено правильно, то лікарі запропонують ще два тести для уточнення тяжкості патології:
- детальне ультразвукове обстеження хребта плода;
- амніоцентез – процедуру, при якій через голку береться невелика кількість навколоплідної рідини і вже потім в ній вимірюється кількість альфа-фетопротеїну.
Що дає діагностування спинно-мозкової кили до народження дитини?
Якщо спинно-мозкова кила діагностована до народження дитини, лікарі можуть доставити матір в спеціально обладнаний центр, де одразу ж після народження дитину піддадуть необхідному лікуванню. Іноді це зменшує наслідки цієї вродженої вади розвитку. Крім того, медики можуть надати батькам необхідну інформацію та підтримку.
Вважається, що проведення пологів шляхом кесарського розтину також може зменшити наслідки спинно-мозкової кили. Отож у лікарів і батьків є час підготуватися до цієї операції.
Переглянуто редакційною колегією I.B.I.S.: 15/02/2002
Дивіться також:
- Спинно-мозкова кила
- Спинномозкова кила (посібник для батьків)
- Аненцефалія
- Енцефалоцеле
- Ініенцефалія
- Роль фолієвої кислоти у запобіганні вадам нервової трубки
- Запобігання вродженим вадам розвитку за допомогою фолієвої кислоти
- Фолієва кислота (Інформація для батьків)
- Фолієва кислота: спеціальне видання
- Фільм про спинно-мозкову килу
- Основи безперервного догляду дітей із Spina Bifida (відео)
- Консервативне ведення розладів сечового міхура та кишечника у дітей із spina bifida (методичні рекомендації)
- Основи безперервного догляду дітей із розщілиною хребта і гідроцефалією (рекомендації для батьків)
Синдром Арнольда-Кіарі
(Arnold-Chiari Malformation)
Т.І. Стеценко
Лікар-невролог
Київської міської дитячої клінічної лікарні №1
Синдром Арнольда-Кіарі – це вада розвитку стовбура головного мозку, при якому є каудальне зміщення довгастого мозку, хробака мозочка, у деяких випадках і моста, подовження порожнини ІV шлуночка у великий потиличний отвір. Хробак мозочка, довгастий мозок, ІV шлуночок розташовані у верхній шийній частині спинномозкового каналу.
Синдром вперше був описаний у 1891 році H. Chiari, у 1895 році V.Arnold.
Залежно від наявності поєднаних вад виділяють декілька видів цієї аномаліі:
- АНОМАЛІЯ АРНОЛЬДА-КІАРІ І – вклинення мигдаликів мозочка через великий потиличний отвір у велику цистерну мозку. У 15%-20% пацієнтів ця вада поєднується з гідроцефалією, а у 50% хворих вада асоціюється з сірінгоміелією.
- АНОМАЛІЯ АРНОЛЬДА-КІАРІ ІІ – відрізняється від І типу більшим ступенем пролапсу мигдаликів мозочку через foramen magnum. Крім мигдаликів мозочка, у велику цистерну та спинно-мозковий канал зміщуються хробак мозочка та довгастий мозок. Тоді ІV шлуночок стає опущеним та подовженим. Ця аномалія асоціюється з мієломенінгоцеле та гідроцефалією. Часто ця вада поєднується з іншими дизрафіями – стенозом водогону мозку, маленькою задньою черепною ямою, недорозвинутим tentorium cerebelli.
- АНОМАЛІЯ АРНОЛЬДА-КІАРІ ІІІ – це варіант потиличного енцефалоцеле, при якому кила пролабує у великий потиличний отвір. При цьому всі структури задньої черепної ями знаходяться як у киловому мішку, так і в спинномозковому каналі.
- АНОМАЛІЯ АРНОЛЬДА-КІАРІ ІV – повна або часткова агенезія хробака мозочка, гіпоплазія мозочка та кистозне розширення ІV шлуночка. Ступінь прояву цих аномалій може бути різною: від часткової відсутності хробака (ще має назву – варіант Денді–Уокера) до майже повної відсутності мозочка та відкриття ІV шлуночка з утворенням кисти.
Клінічна картина:
Діагностика синдрому Арнольда-Кіарі є складною. Провідними серед помилкових діагнозів є – розсіяний склероз, міастенія, психіатричні хвороби. Більшість симптомів пов’язана зі здавленям стовбура мозку та нижніх черепних нервів.
Симптоми:
ГОЛОВНИЙ БІЛЬ – характерний початок із шийної ділянки. Описується як гострий, пульсуючий, переходить з потиличної у лобну ділянку голови і часто позаду або навкруги очей. Біль може провокуватися кашлем, напруженням, чиханням. При нахилі голови вперед біль зменшується, триває від декількох хвилин до декількох годин. Може бути нудота, але блювання не буває.
ДИСФАГІЯ – труднощі при ковтанні як рідкої, так і твердої їжі, поперхування. Швидке прогресування може призвести до аспірації. Зниження глоткового рефлексу.
БІЛЬ У КІНЦІВКАХ – пацієнти скаржаться на біль у шиї або в руці, що часто посилюється при напруженні в руці, втомі, підійманні вантажу. Біль, як правило, однобічний, за характером як тупий, так і гострий, кинжальний, стріляючий. Одночасно може спостерігатись слабкість у руці або порушення координації у ній.
ДИСФОНІЯ – зміна характеру та тембру голосу – це поширена скарга. Частіше помічають оточуючі люди. Також порушення модуляції голосу під час голосного співу або розмови, рідше відзначається ковтання звуків під час розмови.
ПАРЕСТЕЗІІ – відчуття оніміння, поколювання (порушення пропріоцептивної чутливості) часто може супроводжувати біль, переважно у тих же ділянках, що і біль. Відчуття оніміння також завжди однобічне може прогресувати протягом місяців і навіть років, поширюючись на нижні кінцівки та тулуб.
ЗОРОВІ ПОРУШЕННЯ – нечіткість контурів аж до диплопіі, ністагм, частіше при погляді донизу і в бік, можуть бути труднощі в читанні, скотоми, деякі пацієнти не мають скарг на зорові порушення, і ністагм визначають при клінічному огляді.
АТАКСІЯ – порушення ходи у вигляді нестійкості, падіння, зіткнення зі стінками та дверима при ході. Іноді пацієнти відзначають спастику в ногах, яка проявляється при ході.
СІНКОПЕ – непередбачені короткі, тривалістю від 30 секунд до 2 хвилин епізоди втрати свідомості описані в літературі. Після них не буває потьмарення свідомості.
ІНШІ СИМПТОМИ – нудота, запаморочення, дискоординація, тахі-, брадікардія, гіпо-, гіпертензія артеріальна, дзвін у вухах, зниження слуху, депресія, хронічна втома, апное уві сні, проблеми ШКТ, часті сечовиділення, осцилопсія – рідкий симптом – ілюзія коливання нерухомих речей.
Асоційовані синдроми:
Синдром Арнольда-Кіарі часто поєднується з сірінгомієлією (50%), полімікрогірією, гетеротопією, синдромом Кліппеля-Фейля, spina bifida, стенозом водогону мозку, платібазією, порушенням нейрональної міграціі. Дисплазія тенторіуму зустрічається майже завжди, як і маленька задня черепна яма.
80%-90% пацієнтів з цим синдромом мають ще гіпоплазію мозолистого тіла.
Синдром Арнольда-Кіарі також входить до складу декількох хромосомних синдромів: 1q+, 4p-, +8M, +9M, 3q+, 5p+, 9p+, +13, +18 триплоїдія.
Дані обстежень:
АНОМАЛІЯ АРНОЛЬДА-КІАРІ І – при МРТ на середньому сагітальному зрізі (краще на Т1 томограмах) мигдалики мозочка розташовані нижче лініі, яка з’єднує краї клиноподібної та потиличної кісток. Для встановлення діагнозу аномалії пролапс повинен бути не менше 3 мм. Крім ектопії миндаликів, часто спостерігається зміщення стовбура мозку вперед та згладжування моста. Зміщення стовбура призводить до симптому “сходинки”, який проявляється у місці переходу довгастого мозку у спинний.
АНОМАЛІЯ АРНОЛЬДА-КІАРІ ІІ – розташування середини ІV шлуночка нижче лінії, яка з’єднує горбок сідла і випинання на внутрішній поверхні потиличної кістки – головна ознака на МРТ. Окрім того, можуть бути представлені всі дизрафіі.
АНОМАЛІЯ АРНОЛЬДА-КІАРІ ІІІ – енцефалоцеле.
АНОМАЛІЯ АРНОЛЬДА-КІАРІ ІV – описана вище.
Етіологія:
Аномалія Арнольда-Кіарі етіологічно різноманітна і описана при багатьох інших вадах розвитку, хромосомних синдромах.
Ризик для сибсів пробанда – 7%, для нащадків хворих на цю ваду – 7%.
Частота виникнення: 1:200 – 1:500 новонароджених живими.
Співвідношення статі: Ч1 : Ж1.
Патогенез:
Існує декілька теорій розвитку цієї вади. Одна з них подана McLone & Knepper, за якою причиною вклинення стовбура мозку та мозочка у великий потиличний отвір є аномально маленька задня черепна яма при абсолютно нормальному мозочку. За іншою теорією, вада розвивається при зрощенні нижнього відділу медулярної трубки з задньою стінкою спинно-мозкового каналу. У процесі росту не відбувається підтягування нижнього відділу спинного мозку доверху.
Якщо аномалія Арнольда-Кіарі асоціюється з мієломенінгоцеле, то причиною може бути тракція спинного мозку, дистальний кінець якого знаходиться в області килового мішка. Внаслідок вклинення мозку у великий потиличний отвір стискуються отвори Люшка, таким чином розвивається гідроцефалія.
Вік прояву:
Синдром Арнольда-Кіарі може виникнути і прогресувати протягом всього внутрішньоутробного періоду, бути виявленим пренатально, проявитися як з народження (ІІ, ІІІ типи), так і у дорослих.
Диференційний діагноз:
Пухлини задньої черепної ями, краніоспінальні пухлини (зокрема, менінгеома великого потиличного отвору), спіно-церебелярні дегенерації.
Дитинство:
Розвиток дітей з І типом вади може бути нормальним. Взагалі розвиток дітей з вадою Арнольда-Кіарі залежить від супутніх вад головного та спинного мозку та від розвитку гідроцефалії. 15%-20% пацієнтів з вадою І типу можуть мати гідроцефалію.
Лікування:
Оперативне: вентрикулоперитонеостомія та декомпресія стовбура за допомогою ламінектоміі. Крім того:
- лікування поєднаних вад – хірургічна корекція дефекту хребта;
- лікування неврологічних, урологічних, ортопедичних ускладнень;
- запобігання менінгіту;
- важливий контроль за гідроцефалією.
Запобігання:
Медико-генетичне консультування. Аномалію Арнольда-Кіарі ІІ та ІІІ типу можна діагностувати пренатально за допомогою ультразвукового дослідження плоду.
Номер з каталогу МІМ:
207950 Chiari Malformation Type II.
Література:
- McKusick VA. Arnold – Chiari malformation. In: Mendelian Inheritance in Man:Catalogs of Autosomal Dominant, Autosomal Recessive,and X-linked Phenotypes. 10th ed.V.2.Baltimore:The Johns Hopkins University Press, 1992.
- Warkany J. Arnold – Chiari malformation. In: Congenital Malformations: Notes and Comments. Chicago: Yearbook Medical Publishers, Inc., 1971:220.
- Baraitser M. Chiari Type 1. In: The Genetics of Neurological Disoders. Second Edition. Oxford University Press,1990:66-67.
- A.James Barkovich. Arnold – Chiari malformation. In Pediatric Neuroimaging. 1999:238-246.
- D. Bergsma, MD, MPN. Birth Defects Compendium. Published for The National Foundation – March of Dimes. 1979:139, 748.
Переглянуто редакційною колегією I.B.I.S.: 8/05/2003
Сучасна стратегія пренатальної діагностики на ранніх термінах вагітності
(Prenatal Diagnosis in Early Pregnancy)
Зоряна Олексіївна Сосинюк
завідуюча відділенням пренатальної ультразвукової діагностики
Рівненського обласного медико-генетичного центру
Народження хворої дитини пов’язане з важким моральним та матеріальним тягарем як для сім’ї, так і для суспільства. Тому пренатальне виявлення вродженої і спадкової патології продовжує залишатися одним з найактуальніших питань охорони здоров’я. Спеціалісти в галузі допологової діагностики все частіше намагаються перенести обстеження на більш ранні терміни вагітності. Це пояснюється, насамперед, бажанням лікаря раніше виявити патологію, прийняти правильне рішення щодо подальшого супроводу вагітності.
Застосування новітніх технологій, розвиток діагностичної апаратури дозволяють досягти значних успіхів в ранньому виявленні захворювань у плода, використовуючи нові ультразвукові маркери. Розробка та впровадження в практику охорони здоров’я пренатальних скринінгових програм надає можливість не тільки діагностувати патологічний перебіг вагітності, але і формувати групи високого ризику з виникнення хромосомних аномалій у плода.
|
Оптимальним для ехографії в першому триместрі вважається термін вагітності, коли інформація про розвиток ембріона буде максимально повною і точною.
З практики добре відомо що візуалізація плідного мішечка можлива з початку п’ятого тижня вагітності (від першого дня останньої менструації) або з початку третього тижня від моменту запліднення, тобто фактично на стадії імплантації плідного мішечка в ендометрій. Ультразвукове обстеження, проведене на цьому етапі, лише констатує наявність вагітності в порожнині матки чи поза нею, але практично не дає інформації про ембріон.
З початком реєстрації серцевої діяльності ембріона (шостий тиждень від першого дня останньої менструації) при ультразвуковому дослідженні можна достовірно судити про характер вагітності (вагітність розвивається чи завмерла), але анатомія ембріона і в цьому терміні залишається невідомою.
В першому триместрі вагітності не існує магічного терміну найкращої оцінки структур плода. Період ембріогенезу (початок п’ятого – кінець десятого тижня від першого дня останньої менструації) характеризується надзвичайною динамічністю. Відмінною особливістю соноембріології є безперервні якісні морфологічні зміни анатомічних утворів ембріона, активний його ріст. Ехографічне зображення щоденно змінюється. Очевидно, що в цих умовах неможливо проводити ефективну діагностику вад розвитку.
На межі 10-11 тижня від першого дня останньої менструації закінчується формування внутрішніх органів. Після періоду ембріогенезу настає плодовий період розвитку, який характеризується “дозріванням” внутрішніх органів і збільшенням їх розмірів. Незважаючи на те, що після десятого тижня нові анатомічні структури вже не виникають, при проведенні ультразвукового обстеження в ранні терміни (до 14 тижнів) можна в кращому випадку констатувати лише наявність того чи іншого органу, але не оцінювати його будову у зв’язку з його невеликими розмірами.
Приведені вище дані свідчать про те, що більшість органів і систем плода стають більш доступні для візуалізації лише після 12 тижня вагітності. Так, класичне зображення “метелика” при оцінці структур головного мозку можна отримати не раніше 12-13 тижня, зображення дрібних деталей обличчя – після 14-го тижня. Стабільно візуалізувати чотирьохкамерний зріз серця, нирки, сечовий міхур можна з 12-13 тижня, шлунок – не раніше 10-11 тижня і т.д. Слід підкреслити, що мова йде не про ексклюзивні дослідження, а про традиційну ехографію доступну всім вагітним, яку проводять в практичній охороні здоров’я на ультразвукових апаратах середнього класу.
На першому етапі скринінгу мова йде про виявлення лише грубих аномалій розвитку, а не про реальний скринінг вроджених вад розвитку. Більшість вад розвитку на цьому етапі або зовсім недоступні для візуалізації, або потребують підтвердження в більш пізні терміни.
Взаємозв’язок між ВВР плода і терміном її виявлення
(трансабдомінальне сканування)
Аненцефалія. Екзенцефалія. Ініонцефалія |
||||
Неможливість адекватної і надійної оцінки анатомії плода до 12 тижнів та здійснення верифікації діагнозу дозволяє зробити висновок про те, що діагностика вроджених вад розвитку плода не є основною метою першого скринінгового обстеження. Виявлення аномалій розвитку лише доповнення до головного завдання першого етапу скрінінгу – формування серед вагітних групи ризику виникнення вроджених вад розвитку у плода.
|
Одним з відносно нових і універсальних ехографічних маркерів хромосомних аномалій є товщина комірцевого простору. Перші публікації з’явилися в 1992 році, а на IV Всесвітньому конгресі з ультразвукової діагностики в акушерстві та гінекології, який відбувся в жовтні 1994 року в Будапешті був прийнятий термін “nuchal translucency” (“комірцевий простір”) в якості уніфікованої назви цього маркеру. Сьогодні в 43 країнах світу оцінка товщини комірцевого простору у плода використовується при скринінговому ультразвуковому обстеженні в 10-14 тижнів вагітності. Чіткий зв’язок між збільшенням цього параметру і хромосомними абераціями, а також окремими формами вроджених вад розвитку дозволяє виділяти із загального потоку вагітних тих пацієнток, які потребують пренатальної інвазивної діагностики і динамічного контролю.
Товщина комірцевого простору – це ділянка між внутрішньою поверхнею шкіри плоду і зовнішньою поверхнею м’яких тканин в проекції шийного відділу хребта. Для підвищення ефективності скрінінгового обстеження необхідно дотримуватися таких правил:
- оцінка товщини комірцевого простору проводиться в 10-14 тижнів вагітності при числових значеннях куприково-тім’яного розміру плода від 35 до 85 мм;
- вимірювання товщини комірцевого простору здійснюється в сагітальній площині сканування.
Шийна ділянка плода візуалізується при трансабдомінальному обстеженні і є доступною для вимірювання в 95% спостережень. Трансвагінальний доступ для вимірювання слід використовувати лише у випадках, коли оцінка структур плоду затруднена.
|
Частота виявлення розширенного комірцевого простору у плода коливається від 0,5 до 6%. Частота виявлення хромосомних аберацій у плодів з цим ехомаркером в терміні 10-14 тижнів вагітності варіює від 24 до 75% і тому є показом до пренатального каріотипування. При товщині комірцевого простору більше 2,5 мм ризик хромосомних аномалій у плода підвищується в 23,8 рази. Зі збільшенням товщини комірцевого простору зростає частота виявлення хромосомних аномалій, в той же час як при товщині комірцевого простору менше 2,5 мм фактичний ризик трисомії у жінок 35 років і старше нижчий від вікового.
За останні роки в літературі були опубліковані дані про те, що розширення комірцевого простору може відмічатися не лише при хромосомних дефектах у плода, але і при вадах розвитку, діагностика яких потенційно можлива в більш пізні терміни вагітності, зокрема, вроджених вад серця. Враховуючи те, що в ході пренатального скринінгового обстеження діагностується не більше 30% грубих вад серця, проведення цілеспрямованого ехокардіографічного дослідження в 20-24 тижні у плодів, які мали розширенний комірцевий простір в 10-14 тижнів, дозволить значно підвищити діагностику серцевих аномалій.
Частота хромосомних аномалій і вроджених вад серця у плодів
в залежності від товщини комірцевого простору
Особливість візуалізації комірцевого простору (поява не раніше 10 і зникнення після 14 тижня від першого дня останньої менструації) свідчить про транзиторний характер ехомаркеру, що визначає в даному випадку термін проведення першого етапу скринінгу – 10-14 тижнів.
Підвищення ефективності скринінгового обстеження в першому триместрі вагітності можна досягти шляхом розрахунку комбінованого ризику за даними товщини комірцевого простору в поєднанні з даними біохімічного скринінгу. З одного боку він підвищує ефективність ультразвукового скринінгу, а з іншого дозволяє оцінити ризик народження дитини з хромосомною патологією, здійснити диференційований підхід до призначення інвазивних досліджень як молодим матерям, так і жінкам старшої вікової групи.
Найбільш розповсюдженим лабораторним обстеженням в першому триместрі вагітності є визначення двох біохімічних маркерів: бета-субодиниці хоріонічного гонадотропіну (ХГ) та асоційованого з вагітністю плазменного протеїну А (РАРР-А). Хоріонічний гонадотропін з’являється в материнському кровотоці після імплантації ембріона. Його рівень підвищується до 8-го тижня гестації. В нормі з 8-го по 12–ий тиждень вагітності рівень ХГ починає незначно, а з 18-го тижня – значно знижуватися. Рівень ХГ залежить від маси тіла вагітної. Концентрація його знижується із зростанням ваги. При багатоплідній вагітності показники збільшуються вдвічі і більше.
В 1987 році Bogart M.H. і співавторами доведено, що підвищення рівня ХГ в материнській крові часто зустрічається при хромосомній патології плода (зокрема, при хворобі Дауна). Таким чином, ХГ був віднесенний до біохімічних маркерів хромосомних захворювань. Так, при трисомії 13 рівень бета-субодиниці ХГ підвищується, а при трисомії 18, може різко знижуватися.
В 1991 році Brambati B. і співавторами доведено, що асоційований з вагітністю плазменний протеїн А (РАРР-А), який виробляється трофобластом і з’являється в материнській крові на 28-ий день гестації, знижений в першому триместрі вагітності при синдромі Дауна та при інших хромосомних аномаліях плода. З’ясувалось, що найбільш оптимальним терміном для визначення РАРР-А є період з 10 по 12 тиждень вагітності, коли його вміст в материнській крові при трисомії 21 падає більше, ніж у два рази. Ступінь значущості діагностики хромосомної патології при використанні тільки одного маркера складає 42%. Використання обох маркерів (ХГ і РАРР-А) підвищує можливість діагностики хромосомних аберацій до 60%. З врахуванням віку матері, ефективність діагностики зростає до 75%.
Чутливість методу (%) в залежності від комбінації факторів
Вік – 14-22 (30%).
Вік, альфафетопротеїн (AFP) – 14-22 (37%).
Вік, альфафетопротеїн (AFP), хоріонічний гонадотропін (ХГ) – 14-22 (59%).
Вік, альфафетопротеїн (AFP), хоріонічний гонадотропін (ХГ), некон’югований естріол – 14-22 (69%).
Вік, альфафетопротеїн (AFP), хоріонічний гонадотропін (ХГ), некон’югований естріол, інгібін А – 14-22 (76%).
Вік, комірцевий простір (NT), хоріонічний гонадотропін (ХГ), плазмений протеїн А (PAPP – A) – 11-13 (80-90%).
Крім того, десятки публікацій останніх років показали, що застосування імунобіохімічних маркерів та ультразвукового вимірювання товщини комірцевого простору у плода підвищують діагностичну точність такого підходу, сумарна чутливість якого з індивідуальним врахуванням фактору віку матері, перевищує 90%.
Ризик народження дитини з синдромом Дауна
Після отримання результатів всіх обстежень для кожної жінки з допомогою комп’ютерної програми розраховується індивідуальний комбінований ризик народження дитини з хромосомною патологією. Якщо у вагітної цей ризик перевищує порогове значення (1:250), жінка потрапляє до групи високого генетичного ризику і їй рекомендується проведення інвазивних процедур (в першому триместрі вагітності – біопсія хоріону, в другому триместрі – амніоцентез з метою визначення каріотипу плоду).
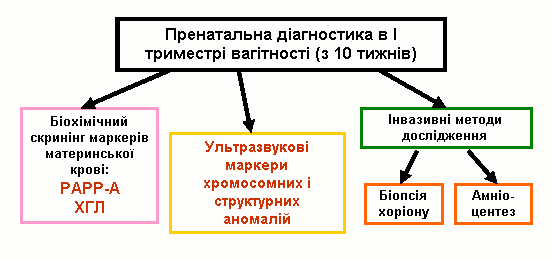
Комплексна пренатальна діагностика в першому триместрі вагітності має очевидні переваги для пацієнтів та клініцистів:
- Формування “групи ризику” серед вагітних щодо вроджених вад розвитку плоду.
- Зменшення кількості інвазивних процедур.
- Зменшення кількості ускладнень інвазивних втручань (“спровоковані викидні“).
- Зменшення вартості допологової діагностики.
Список літератури:
- Бахарев В.А., Каретникова Н.А., Полестеров Ю.А. и др. Значение ранннего амниоцентеза для пренатальной диагностики в І триместре беременности //Акушерство и гинекология.- 1991.- № 4.- С. 9-10.
- Медведев М.В., Алтынник Н.А. Воротниковое пространство у плодов в ранние сроки беременности: новые аспекты пренатальной диагностики //Ультразвуковая діагностика в акушерстве, гинекологии и педиатрии.- 1999.- Т. 7, № 1.- С. 19-26.
- Медведев М.В., Мальмберг О.Л. Новые пренатальные эхографические маркеры хромосомной патологии //Ультразвуковая диагностика в акушерстве, гинекологии и педиатрии.- 1995.- № 1.- С. 13-21.
- Пренатальная диагностика врожденных пороков развития в ранние сроки беременности /Под ред. М.В. Медведєва.- М.: Реальное время, 2000.- 160 с.
- Снайдерс Р.Дж.М., Никалаидес К.Х. Ультразвуковые маркеры хромосомных дефектов плода.- М.: Видар, 1997.- 92 с.
- Юдина Е.В., Медведев М.В. Дородовая диагностика синдрома Дауна в России в 2005 году, или Перинатальная драма в трех частях с прологом и эпилогом //Журнал “Пренатальная диагностика”.- 2007.- № 4.- С.252-257.
- Cucle HS,Van Lith JMM. Appropriate biochemical parameters in first-trimester screening for Down syndrome. Prenat Diagn 1999;19:505-512.
- Nicolaides KH, Sebire NJ, Snijders JM. The 11-14 week scan: The diagnosis of fetal abnormalities. NY, L.The Parthenon Publ. Gr. 1999.
Переглянуто редакційною колегією I.B.I.S.: 18/03/2008
Спинно-мозкова кила
(Spina Bifida)
лікар-генетик
пологового будинку
лікар-генетик
Волинського обласного дитячого
територіального медичного об’єднання
Щороку spina bifida (у перекладі – “розчеплений хребет”, або спинно-мозкова кила) та аненцефалія – дві найбільш поширені форми вад невральної трубки (ВНТ) – вражають понад 300000 новонароджених в усьому світі. Відомі ще з античних часів ці недуги зумовлюють важкі ураження центральної нервової системи з подальшим формуванням інвалідності та смертності. Останні десятиріччя минулого століття ввійшли в історію медицини як час “прориву”, досягнення швидкого та значного прогресу, насамперед у сфері запобігання цим вадам.
За визначенням журналу Американської медичної асоціації (JAMA, 1993): “Одним з найвизначніших винаходів другої половини ХХ століття в медицині є відкриття того факту, що фолієва кислота, простий і широкодоступний водорозчинний вітамін, може запобігти виникненню spina bifida й аненцефалії”.
Spina bifida – результат порушення процесів нейруляції (первинної та вторинної) на ранніх етапах ембріогенезу (до 25-27 днів після зачаття, тобто ще до того, як жінка дізнається, що вона вагітна) під впливом генетичних факторів та деяких тератогенних чинників як фізичної (гіпертермія), так і хімічної природи (гіпервітаміноз А, вальпроєва кислота, антагоністи фолієвої кислоти, гіперглікемія та інші).
В процесі раннього ембріонального розвитку особливе місце належить нейруляції – поетапному формуванню нервової трубки в результаті злиття нервової складки. Закриття переднього відділу відбувається приблизно на 25-ту добу ембріонального розвитку, а каудального (заднього) – на 27-му добу. З цього моменту нейруляція вважається завершеною, і центральна нервова система представлена закритою трубчастою структурою з вузькою каудальною частиною – спинним мозком і значно ширшою головною ділянкою, яка характеризується кількома мозковими пухирями.
Класифікація:
Ембріологічно та з урахуванням типу ураження нервової тканини spina bifida класифікується на:
- відкриту – виникає внаслідок порушення первинної нейруляції, невральна тканина залишається відкритою, або вкрита лише мембраною, часто поєднується з гідроцефалією, мальформацією Арнольда-Кіарі;
- закриту (spina bifida occulta) – зумовлена дефектом вторинної нейруляції, нервова тканина не зачеплена, дефект повністю епітелізований, проте шкірні покриви у ділянці дефекту можуть бути диспластичними.
Відкриті форми spina bifida:
1. Spina bifida cystica uverta – відкрита розщілина хребта з формуванням кистозної спинно-мозкової кили. Відкриті кистозні розщеплення хребта (істинні спинно-мозкові кили) залежно від залучення в патологічний процес нервових структур розділяють на:
- Менінгоцеле – розщеплення хребта з випинанням у дефект твердої мозкової оболонки, але без залучення нервових структур.
- Менінгорадикулоцеле – розщеплення хребта з випинанням в дефект оболонок спинного мозку і його корінців, які частково можуть закінчуватися в стінці мішка, частково входити в нього, створюючи петлю, але в подальшому, поширюючись в міжхребцеві отвори, формують нормальні нерви.
- Менінгомієлоцеле або менінгомієлорадикулоцеле – розщеплення хребта із залученням в кильовий мішок оболонок, спинного мозку і його корінців.
- Мієлоцистоцеле – відносно рідкісна форма спинно-мозкових кил, при яких кінцевий відділ спинного мозку різко розширений за рахунок центрального каналу спинного мозку.
- Spina bifida complicata – ускладнена форма, характеризується поєднанням однієї з вищенаведених форм спинно-мозкових кил із доброякісними пухлинами (ліпомами, фібромами), які фіксовані до оболонок, спинного мозку і його корінців.
2. Rhachischisis posterior (totalis et partialis) – незарощення хребта і його тканин із несформованим спинним мозком – крайній ступінь вади, що ніколи не супроводжується кистозним компонентом і випинанням утворення над шкірою.
3. Дуже рідко (менше 1% випадків) незарощення формується на передньобоковій поверхні каналу і виникають передні спинно-мозкові кили.
Розміщення спинно-мозкових кил по довжині хребта в 90% випадків обмежується попереково-крижовою ділянкою. Грудна та шийна локалізація кил трапляється рідше. При дослідженні матеріалу спонтанних абортів виявлено частіше порушення формування хребта і спинного мозку в грудному та шийному відділах, а також високу частоту дефектів, що охоплюють весь спинний стовбур. Це говорить про те, що ембріон та плід з грубим дефектом формування невральної трубки, як правило, гине.
Spina bifida occulta, яка є найлегшою формою спинальної дизрафії, досить рідко включається у дослідження ВНТ, оскільки часто залишається недіагностованою і відсутнє точне визначення її зв’язку із відкритою spina bifida.
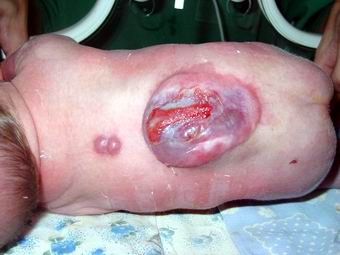
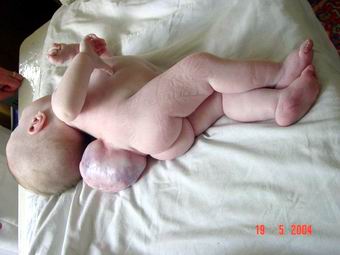
Частота:
ВНТ належать до найпоширеніших вроджених вад розвитку (від 1:300 до 1:2000), однак на їх частоту впливають географічні, етнічні відмінності, застосовувані у системі громадського здоров’я підходи до профілактики. Високі рівні спостерігаються в Індії, Північному Китаї (3,7:1000), Ірландії.
Частота ВНТ є вищою серед європейців, ніж серед африканців, індіанців, японців. Окрім того, протягом останніх десятиріч спостерігається тенденція до зниження ВНТ у ряді країн світу.
Клінічна характеристика:
Прихована щілина хребта звичайно локалізується в поперековому та крижовому відділах. Дефект прикритий незміненими м’язами та шкірою. Зовнішніми ознаками розщелини хребта можуть бути гіпертрихоз, телеангіектазії, ліпоми, дермальні та пілонідальні синуси в ділянці дефекту хребта. Дермальний синус – це вузький хід діаметром 1-2 мм, який йде від поверхні тіла між хребцями до каналу хребта, іноді проникає через тверду мозкову оболонку. Пілонідальний синус звичайно розміщений в м’яких тканинах крижової ділянки і рідко проникає в канал хребта.
Зазвичай, пацієнти з прихованою щілиною хребта не мають нервово-м’язевих порушень. Дефект хребта виявляється як випадкова рентгенологічна знахідка.
Проте слід пам’ятати, що асиметрія ніг та/чи ступнів, сколіоз, слабкість у дистальних відділах кінцівок, у дорослих – больовий синдром, епізоди порушення чутливості можуть бути ознаками прихованої щілини хребта.
Менінгоцеле найчастіше зустрічається в поперековому відділі хребта. Іноді зустрічаються парези та втрати чутливості в нижніх кінцівках; поєднання менінгоцеле з гідроцефалією буває рідко. На шкірі, що покриває киловий мішок, часто бувають телеангіектазії та гіпертрихоз.
Менінгомієлоцеле є причиною неврологічної патології, яка проявляється порушенням функції моторних нейронів спинного мозку, сфінктерів ануса та сечового міхура, відсутністю рефлексів і чутливості залежно від рівня розташування кили.
Менінгомієлоцеле часто асоціюється з вадою Арнольда-Кіарі, яка являє собою вклинення довгастого мозку та мигдаликів мозочка у великий потиличний отвір внаслідок тракції спинного мозку, дистальний кінець якого знаходиться в області кильового мішка. Вада Арнольда-Кіарі може виникнути і прогресувати протягом всього внутрішньоутробного періоду. Симптоматика мальформації Арнольда-Кіарі II у новонароджених включає стридор із паралічем голосових зв’язок, центральне апное, аспірацію, дисфагію, гіпотонію, прогресуючу дисфункцію стовбура головного мозку, мієлопатію, тетрапарез, труднощі із ковтанням, погане смоктання, прогресуючий ністагм, страбізм.
Приблизно у 20% дітей із spina bifida також спостерігаються й інші вроджені вади центральної нервової системи.
Хромосомні аномалії, мутації одного гена та тератогенні впливи визначаються менше ніж у 10% хворих немовлят.
Відомий ряд моногенних синдромів, до складу яких входить spina bifida:
183802 Ектродактилія з обструктивною уропатією, spina bifida та діафрагмальною килою
301410 Спинно-мозкова кила, Х-зчеплена
193500 Синдром Ваарденбурга, тип 1
257920 Окуло-палато-скелетний синдром
169550 Тазово-плечова дисплазія
102510 Акро-пекто-вертебральна дисплазія
601634 Вади невральної трубки, фолат-чутливі
Лікування:
Лікування передбачає хірургічну корекцію дефекту хребта та супутніх уражень центральної нервової системи (шунтування при гідроцефалії, оскільки приблизно у 20% пацієнтів із мієломенінгоцеле при народженні спостерігається значна гідроцефалія; у інших 60–70% вона розвивається після закриття мієломенінгоцеле; у певних пацієнтів встановлення шунта одночасно із закриттям мієломенінгоцеле є цілком обгрунтованим), лікування неврологічних, урологічних, ортопедичних ускладнень.
Пренатальна діагностика:
Пренатальна діагностика проводиться шляхом ультрасонографії, у складних діагностичних випадках можливе використання магнітно-резонансної томографії (МРТ), а також визначення рівня альфа-фетопротеїну в сироватці крові жінки у ІІ триместрі вагітності.
Прогноз:
Сприятливий для пацієнтів з мінімальними неврологічними порушеннями та без гідроцефалії. Прогноз в інших випадках залежить від ступеню ураження ЦНС, наявних ускладнень та супутньої патології. 10% дітей із spina bifida мають затримку розумового розвитку.
Визначення довготермінового прогнозу також є досить важким завданням через покращання лікування деяких медичних ускладнень (наприклад, ниркова недостатність), які раніше значно збільшували ризик смерті у дорослих зі spina bifida. Два дослідження підтверджують виживання майже до третього десятиліття у 52% та 68% пацієнів, у яких дефект був виправлений хірургічним шляхом одразу після народження.
Немовлята зі spina bifida, які виживають, швидше за все, будуть страждати на важкі порушення протягом усього життя, а також існує ризик їх психо-соціальної непристосованості. Медичні проблеми часто виникають внаслідок неврологічного дефекту або його виправлення (наприклад, параліч, гідроцефалія, синдром Арнольда-Кіарі ІІ типу, ендокринні порушення, сирингомієлія), або можуть виникати в наслідок неврологічного дефіциту (наприклад, деформації кінцівок і хребта, порушення функції сечового міхура, кишківника, сексуальна дисфункція та проблеми із навчанням).
Водночас із моральними втратами існують також і вражаючі матеріальні витрати. Тільки у Сполучених Штатах загальна вартість spina bifida протягом життя (прямі витрати на медичні, розвиткові та освітні послуги, та непрямі внаслідок захворювання чи смерті) для хворих немовлят у 1988 р. склали 500 млн. доларів, або 294 тис. на кожного новонародженого (An assessment of total costs and policy implications. In: Waitzman NJ, Scheffler RM, Romano PS. The cost of birth defects: estimates of the value of prevention. Lanham, Md.: University Press of America, 1996:145-77.).
Запобігання:
Зв’язок між харчуванням та ВНТ є потужним інструментом первинного запобігання. Після публікації результатів двох рандомізованих досліджень (MRC Vitamin Study Research Group. Prevention of neural tube defects: results of the Medical Research Council Vitamin Study. Lancet 1991;338:131-137 [Medline]. Czeizel AE, Dudas I. Prevention of the first occurrence of neural-tube defects by periconceptional vitamin supplementation. N Engl J Med 1992;327:1832-1835 [Medline]) Службою Громадського Здоров’я США у 1992 р. була видана рекомендація всім жінкам, здатним завагітніти, вживати 400 мкг фолієвої кислоти щоденно.
Дешевим методом збільшення споживання фолієвої кислоти, що не потребує зміни поведінки, є додавання фолієвої кислоти до широковживаних продуктів – фортифікація продуктів харчування. Враховуючи те, що більшість вагітностей є незпланованими, збагачене фолієвою кислотою борошно є найефективнішим методом запобігання ВНТ і попередження смертності кількох сотен українських дітей щорічно.
Понад 28 країн світу запровадили збагачення пшеничного борошна фолієвою кислотою. Серед цих країн США, Канада, Аргентина, Чилі, Казахстан тощо. Ще 12 країн перебувають на стадії запровадження фортифікації.
Збагачення зернових продуктів фолієвою кислотою допоможе заощадити значні кошти за рахунок попередження ВНТ у плода. Так, вже тепер в США це дозволяє заощаджувати 94 млн.доларів щорічно.
Нові завдання та перспективи:
Прогрес у клінічному лікуванні вад невральної трубки, дослідження патогенезу та підходи щодо первинного попередження відкривають нові простори та висувають суттєві завдання. Сьогодні нагальною проблемою, з якою стикаються спеціалісти з медицини та охорони здоров’я є те, як впровадити наші знання щодо первинного попередження у практику. Кожен день народжуються діти із вадами, вагітні жінки вибірково переривають вагітність.
Можливі варіанти попередження включають фортифікацію (збагачення) борошна фолієвою кислотою, збільшення споживання їжі з добавками фолієвої кислоти та збільшення прийому вітамінів, що містять фолієву кислоту. Вдосконалення знань, зміна звичок жінок та медичних спеціалістів дуже важливі у намаганнях зрозуміти весь превентивний потенціал фолієвої кислоти. Всесвітнє медичне співтовариство повинно вдатися до погоджених дій для вирішення цієї проблеми.
Брюс Пейн, британський актор театру і кіно, який страждає на легку форму spina bifida. Фільм “One Point O” з Брюсом Пейном у 2003 році отримав схвальні відгуки на Канському кінофестивалі. |
Література:
- Спинно-мозкова кила /І.Р. Бариляк, Ю.О. Орлов, О.А. Данилов, В.О. Пирогов, Г.В. Скибан та ін.– К., 2003. – 101 с.
- Adzick NS, Sutton LN, Crombleholme TM, et al: Successful fetal surgery for spina bifida. Lancet 1998 Nov 21; 352(9141): 1675-6.
- Aubry MC, Aubry JP, Dommergues M: Sonographic prenatal diagnosis of central nervous system abnormalities. Childs Nerv Syst 2003 Aug; 19(7-8): 391-402.
- Botto et all. Medical Progress: Neutral-Tube Defects, N Engl J Med 1999; 341: 1509-1519, Nov 11, 1999.
- CDC: Economic burden of spina bifida–United States, 1980-1990. MMWR Morb Mortal Wkly Rep 1989 Apr 21; 38(15): 264-7.
- Czeizel AE, Dudas I: Prevention of the first occurrence of neural-tube defects by periconceptional vitamin supplementation. N Engl J Med 1992 Dec 24; 327(26): 1832-5.
- Lemire RJ, Beckwith JB, Warkany J: Anencephaly. NY: Raven Press; 1978: 1-271.
- Lindhout D, Omtzigt JG: Teratogenic effects of antiepileptic drugs: implications for the management of epilepsy in women of childbearing age. Epilepsia 1994; 35: S19-28.
- Mills JL, Conley MR: Periconceptional vitamin supplementation to prevent neural tube defects: how can we do it? Eur J Obst Gynecol Reprod Biol 1995; 61: 49-55.
- MRC Vitamin Study Research Group: Prevention of neural tube defects: results of the Medical Research Council Vitamin Study. Lancet 1991 Jul 20; 338(8760): 131-7.
- Oakley GP, Mandel JS. Folic acid fortification remains an urgent health priority. BMJ. 2004 Dec 11;329(7479):1376.
- Richard G Ellenbogen. Neural Tube Defects in the Neonatal Period.
(http://www.emedicine.com/ped/topic2805.htm) - Sever LE. Use of folic acid supplementation in the periconceptional period provides great promise for the primary prevention of the neural tube defects (NTDs), anencephaly, and spina bifida.J Nurse Midwifery. 1992 Sep-Oct;37(5):350.
- Shurtleff DB, Lemire RJ: Epidemiology, etiologic factors, and prenatal diagnosis of open spinal dysraphism. Neurosurg Clin N Am 1995 Apr; 6(2): 183-93.
- Warkany Joseph. Congenital Malformations: Notes and Comments. Year Book Medical Publishers Inc. 1971: 272-291.
- Yen IH, Khoury MJ, Erickson JD: The changing epidemiology of neural tube defects. United States, 1968- 1989. Am J Dis Child 1992 Jul; 146(7): 857-61.
Переглянуто редакційною колегією I.B.I.S.: 18/11/2005
Дивіться також:
- Спинно-мозкова кила (Інформація для батьків)
- Спинномозкова кила (посібник для батьків)
- Аненцефалія
- Енцефалоцеле
- Ініенцефалія
- Роль фолієвої кислоти у запобіганні вадам нервової трубки
- Запобігання вродженим вадам розвитку за допомогою фолієвої кислоти
- Фолієва кислота (інформація для батьків)
- Фолієва кислота: спеціальне видання
- Фільм про спинно-мозкову килу
- Основи безперервного догляду дітей із Spina Bifida (відео)
- Консервативне ведення розладів сечового міхура та кишечника у дітей із spina bifida (методичні рекомендації)
- Основи безперервного догляду дітей із розщілиною хребта і гідроцефалією (рекомендації для батьків)
Ініенцефалія
(Iniencephaly)
О.Ю. Суботяк
Лікар-невролог
Рівненської обласної дитячої лікарні
Включення:
- Незавершене формування основи черепа, особливо в ділянці великого потиличного отвору.
- Рахішизис.
- Патологічне посилення лордозу хребта.
Основні діагностичні критерії:
Ініенцефалія – відсутність частини або усієї потиличної кістки зі значним розширенням великого потиличного отвору (inion – потилиця), внаслідок чого значна частина головного мозку розміщена в ділянці черепної ямки та частково у верхньому відділі хребетного каналу, хребці якого не мають дужок та остистих відростків.
Клінічні ознаки:
Голова по відношенню до короткого тіла велика. Обличчя повернене догори. Шия різко вкорочена або відсутня. Шкіра обличчя зливається зі шкірою передньої грудної стінки, а скальп (якщо він присутній) поєднаний зі шкірою спини. Волосся на тімені відсутнє та надмірно поширене на шиї і спині. Деформована і ретрофлексована потилиця може бути з’єднана з нижнім відділом хребта. У більшості випадків присутній рахішизис, але деформовані спинний мозок та хребет покриті мозком, мозочком і шкірою дорсально так, що на поверхні не видно відкритої нервової пластинки і хребта – це є закрита ініенцефалія. Явно виражена ретрофлексія голови також часом з’являється у ініенцефалів чи у плодів з відкритим рахішизисом (відкрита ініенцефалія). При закритій ініенцефалії, складові черепа залишаються всередині черепної коробки, а при відкритій формі, вони випинаються у енцефалоцеле чи виявляються, як екзенцефалія чи аненцефалія. Хребет завжди сильно деформований та вкорочений, особливо його шийний відділ, через формування блокади хребта і дефіцит хребців.
Асоційовані вади:
Аненцефалія, енцефалоцеле, гідроцефалія, циклопія, рахішизис, з’єднання або дефіцит ребер, діафрагмальна кила, порушення поділу на долі легень, одна пуповинна артерія, омфалоцеле, транспозиція (внутрішніх) органів, підковоподібна і полікистозна нирка, неперфорований анус, клишоногість, деформовані вуха, незарощене піднебіння і розщеплена губа.
Диференційний аналіз:
Ініенцефалію потрібно диференціювати з:
- Синдромом Кліппеля-Фейля, для якого характерно:
- вкорочення шиї за рахунок злиття шийних хребців;
- менінгоцеле шийного відділу;
- сумісність з постнатальним життям.
- Аненцефалією, що характеризується відсутністю:
- великого мозку;
- кісток склепіння черепа;
- м’яких тканин склепіння черепа.
У випадку ініенцефалії м’які тканини та кістки склепіння черепа збережені. Головний мозок також збережений, але можуть спостерігатися макро- і полігірія, а також порушення цитоархітектоніки кори великих півкуль.
Етіологія:
Є повідомлення про зв’язок розвитку ініенцефалії у плода вагітної з сифілісом, а також з прийомом седативних препаратів.
Той факт, що ініенцефалію можна викликати експериментально при вживанні вагітною наркотиків, свідчить про те, що існує можливість середовищного спричинення хвороби.
У тварин захворювання викликали введенням їм вінбластину, стрептонігрину, трипаранолу.
Частота:
Точно не встановлена. Наприклад, 1:896 в Англії, 1:65 в Індії. Ініенцефалія у сибсів зустрічається частіше.
Співвідношення статей:
Співвідношення між статями складає 0,28.
Патогенез:
Відомо декілька гіпотез розвитку захворювання. До найбільш розповсюджених відноситься персистуючий шийний лордоз ембріона на 3-му тижні його розвитку, що призводить до порушення закриття невральної трубки. А також гіпотеза аномального розвитку ростральної частини хорди та шийно-потиличної ділянки.
Вік прояву: пренатальний період.
Пре/неонатальний періоди:
Вагітність часто триває повний термін і ускладнюється гідрамніоном. Внаслідок викривлення тіла плода порушення його положення (таке як, наприклад, сідничне, ніжне, лицеве чи підборідкове) є досить частим і часом небезпечним для життя матері.
Більшість ініенцефалів є карликами при народженні. Ступінь затримки внутрішньоутробного розвитку важко оцінити, бо лише у декількох випадках гестаційний вік та вага були записані. У одному з описаних випадків пологи відбувались на восьмому місяці, і вага плоду була лише 1470 грамів.
Акушерська тактика:
До досягнення життєздатності плода жінці необхідно запропонувати переривання вагітності. Якщо достовірний діагноз встановлено в більш пізньому терміні, рекомендується консервативне ведення пологів.
Прогноз:
Ініенцефалія практично завжди закінчується смертю у неонатальному періоді.
Літературні джерела:
- Ромеро Р., Пилу Дж. и др. Пренатальная диагностика врожденных пороков развития плода /Пер. с англ.- М.: Медицина,1994.- С. 74-75.
- Тератология человека /Под. ред. Г.И. Лазюка. – М.: Медицина, 1979, C. 106.
- Jones KL. Smith’s Recognizable Patterns of Human Malformation. 5-th ed. 1981.
- Warkany J. Congenital malformation (notes and comments).- P. 234-236.
Переглянуто редакційною колегією I.B.I.S.: 2/02/2002
Дивіться також:
- Аненцефалія
- Енцефалоцеле
- Спинно-мозкова кила (Spina Bifida)
- Спинно-мозкова кила (Spina Bifida) (інформація для батьків)
- Спинномозкова кила (посібник для батьків)
- Фолієва кислота (інформація для батьків)
- Фолієва кислота (спеціальне видання)
- Фільм про спинно-мозкову килу
- Основи безперервного догляду дітей із Spina Bifida (відео)
- Роль фолієвої кислоти у запобіганні вадам нервової трубки
- Консервативне ведення розладів сечового міхура та кишечника у дітей із spina bifida (методичні рекомендації)
- Основи безперервного догляду дітей із розщілиною хребта і гідроцефалією (рекомендації для батьків)
Енцефалоцеле
(Encephalocele)
Н.І. Юськів
Лікар-генетик
Волинського обласного дитячого територіального медичного об’єднання
Включені:
- Краніальне менінгоенцефалоцеле
- Розщелина черепа
Виключені:
- Гідроцефалія
- Меккеля синдром
- Менінгоцеле
- Орбітальне цефалоцеле
- Асоціації дизрафій
Основні діагностичні критерії:
- Килоподібне випинання тканини головного мозку та його оболонок через дефекти кісток черепа
Клінічні ознаки:
Енцефалоцеле звичайно локалізується у висковій, лобній ділянках, біля кореня носа, біля внутрішнього кута ока, іноді в області носоглотки, та найчастіше (близько 70% випадків) зустрічається у потиличній області. Великі черепно-мозкові кили супроводжуються важкими неврологічними розладами. Дефект кісток черепа підтверджується рентгенографічно.
При потиличній черепно-мозковій килі тканини мозку та його оболонок випинаються через дефект потиличної кістки та розширений for. magnum. Іноді кила сягає великих розмірів і може поєднуватися з розщелиною хребта в цервікальному відділі.
Ускладнення:
При порушенні цілості шкірних покривів енцефалоцеле може ускладнитися менінгітом. Необережні маніпуляції в області носоглотки при наявності там черепно-мозкової кили теж можуть призводити до інфікування.
Знахідки:
Потилична черепно-мозкова кила є причиною порушення циркуляції спинно-мозкової рідини, що призводить до розвитку гідроцефалії у більше, ніж 50% випадків. Енцефалоцеле може зустрічатись як ізольована вроджена вада розвитку, або ж з іншими аномаліями входити до складу різних синдромів.
Патогенез:
Основою даного церебрального дефекту є порушення змикання невральної трубки по середній лінії з протрузією мозку.
Співвідношення статі: Ч1 : Ж1.
Частота:
Оцінюється як 1 на 2000 живих новонароджених. Найвища частота в Ірландії. Надзвичайно часто зустрічається в Таіланді.
Повторний ризик для сибсів:
Ризик повторного народження хворої дитини становить 6%.
Генне картування і групи зчеплення генів: невідомі.
Профілактика:
Рекомендоване медико-генетичне консультування. Преконцепційне застосування та вживання в першому триместрі вагітності фолієвої кислоти зменшує ризик виникнення у плода дефектів змикання невральної трубки.
Лікування:
Необхідна якнайшвидша хірургічна корекція дефекту.
Прогноз:
Приблизно 20% дітей мають розумову відсталість та інвалідність в зв’язку з неврологічними розладами.
Діагностика носійства: невідома.
Список літератури:
- Cullen MT, Athanassiadis AP, Romero R. Prenatal Diagnosis of Anterior Parietal Encephalocele with Transvaginal Sonography. Obstetrics and Gynecology 1990;75:489-491.
- Fisher NL, Smith DW. Occipital encephalocele and early gestational hyperthermia. Pediatrics 1981;4:480-483.
- Gorlin RJ, Cohen MM, Levin LS. Encephalocele. In: Syndromes of the Head and Neck. 3d ed. Oxford: Oxford University Press, 1990:568-570.
- Guthkelch AN. Occipital cranium bifidum. Archives of Disease in Childhood 1970;45:104-109.
- Lorber J. The prognosis of occipital encephalocele. Developmental Medicine and Child Neurology 1967;13:75-83.
- Shapiro K. Encephalocele. In: Buyse ML, ed. Birth Defects Encyclopedia. Dover: Center for Birth Defects Information Services, Inc., 1990:614-615.
- Warkany J. Encephalocele. In: Congenital Malformations: Notes and Comments. Chicago: Yearbook Medical Publishers, Inc., 1971:211-216.
Переглянуто редакційною колегією I.B.I.S.: 15/02/2002
Дивіться також:
- Аненцефалія
- Ініенцефалія
- Спинно-мозкова кила (Spina Bifida)
- Спинно-мозкова кила (Spina Bifida) (інформація для батьків)
- Спинномозкова кила (посібник для батьків)
- Фолієва кислота (інформація для батьків)
- Фолієва кислота (спеціальне видання)
- Фільм про спинно-мозкову килу
- Основи безперервного догляду дітей із Spina Bifida (відео)
- Роль фолієвої кислоти у запобіганні вадам нервової трубки
- Консервативне ведення розладів сечового міхура та кишечника у дітей із spina bifida (методичні рекомендації)
- Основи безперервного догляду дітей із розщілиною хребта і гідроцефалією (рекомендації для батьків)
Аненцефалія
(Anencephaly)
Наталія Ігорівна Юськів
Лікар-генетик
Волинського обласного дитячого територіального медичного об’єднання
Основні діагностичні критерії:
- Відсутність великого мозку, кісток склепіння черепа і м’яких тканин.
Основні клінічні ознаки:
Великий мозок відсутній. На місці мозкової речовини розміщена багата на кровоносні судини сполучна тканина з кистозними порожнинами, яка може імітувати звивини і борозни. Черепні нерви рудиментарні, хоча периферична нервова система добре розвинена. Характерні мікромієлія та деформації спинно-мозкового каналу.
Аненцефалія часто поєднується із spina bifida. При краніорахішизисі васкуляризована сполучна тканина заповнює дефекти черепа і хребта. Шия коротка, обличчя розвернуте догори за рахунок вираженого лордозу цервікального відділу хребта, вуха торкаються плечей. Повіки та очні м’язи, як правило, розвинуті добре. Ніс широкий, язик великий. Часто відмічається розщелина піднебіння. Кістки склепіння черепа відсутні, орбіти мілкі, що є причиною протрузії очних яблук.
Діти з даною вадою розвитку народжуються мертвими або ж помирають в перші години чи дні життя.
Етіологія:
Мультифакторіальна. Причиною виникнення ізольованих дизрафій (в т.ч. аненцефалії) є одночасна взаємодія як генетичних факторів, так і факторів зовнішнього середовища (наприклад гіпертермія у матері в 1-му триместрі вагітності, вплив токсичних речовин довкілля, материнсько-плодові інфекції, вживання антагоністів фолієвої кислоти).
Патогенез:
Незмикання невральної трубки, рідше – її розрив в ембріональному періоді.
Співвідношення між статтю: Ч 1 : Ж 3-7
Популяційна частота: 1 на 1000 живих новонароджених.
Вік маніфестації:
Аненцефалію можна діагностувати пренатально, застосовуючи ультрасонографію та деякі лабораторні дослідження, а саме: визначення рівня альфа-протеїну в сироватці крові матері та навколоплідних водах.
Асоційовані знахідки:
Відсутність гангліозних клітин сітківки, аплазія нейрогіпофізу, наднирників.
Лікування: немає.
Прогноз: несприятливий.
Діагностика носійства: невідома.
Профілактика:
Рекомендоване медико-генетичне консультування. Відомо, що преконцепційне застосування та вживання в 1-му триместрі вагітності (період органогенезу) фолієвої кислоти, значно знижує ризик виникнення у плода дефектів змикання невральної трубки.
Диференційний діагноз:
- Гідраненцефалія
- Асоціації дизрафій
- Арнольда-Кіарі мальформація (мозок).
Номер з каталогу МІМ:
206500 Anencephaly
Список літератури:
- Brock DJH, Sutcliffe RG. Alpha-Fetoprotein in the Antenatal Diagnosis of Anencephaly and Spina Bifida. Lancet 1972;2:197-199.
- Golden JA, Chernoff GF. Multiple Sites of Anterior Neural Tube Closure in Humans: Evidence from Anterior Neural Tube Defects (Anencephaly). Pediatrics 1995;95:506-510.
- Gorlin RJ, Cohen MM, Levin LS. Anencephaly. In: Syndromes of the Head and Neck. 3d ed. Oxford: Oxford University Press, 1990:565-567.
- Kurent JE, Sever JL. Perinatal Infections and Epidemiology of Anencephaly and Spina Bifida. Teratology 1973;8:359-362.
- McKusick VA. Anencephaly. In: Mendelian Inheritance in Man: Catalogs of Autosomal Dominant, Autosomal Recessive, and X-Linked Phenotypes. 10th ed. V. 2. Baltimore: The Johns Hopkins University Press, 1992:1219.
- Shapiro K. Anencephaly. In: Buyse ML, ed. Birth Defects Encyclopedia. Dover: Center for Birth Defects Information Services, Inc., 1990:139-140.
- Warkany J. Anencephaly. In: Congenital Malformations: Notes and Comments. Chicago: Yearbook Medical Publishers, Inc., 1971:189-199.
Переглянуто редакційною колегією I.B.I.S.: 15/02/2002
Дивіться також:
- Енцефалоцеле
- Ініенцефалія
- Спинно-мозкова кила (Spina Bifida)
- Спинно-мозкова кила (Spina Bifida) (інформація для батьків)
- Спинномозкова кила (посібник для батьків)
- Фолієва кислота (інформація для батьків)
- Роль фолієвої кислоти у запобіганні вадам нервової трубки
- Роль фолієвої кислоти у запобіганні розщілини губи і/або піднебіння
- Фолієва кислота (спеціальне видання)
- Фільм про спинно-мозкову килу
- Основи безперервного догляду дітей із Spina Bifida (відео)
- Запобігання вродженим вадам розвитку за допомогою фолієвої кислоти
- Консервативне ведення розладів сечового міхура та кишечника у дітей із spina bifida (методичні рекомендації)
- Основи безперервного догляду дітей із розщілиною хребта і гідроцефалією (рекомендації для батьків)
Анемія Мінковського-Шоффара
(Spherocytosis Hereditary)
Лікар-неонатолог
Волинського обласного дитячого територіального медичного об’єднання
Визначення:
Генетичний дефект білка мембрани, утворення сфероцитів, посилений їх гемоліз в селезінці.
Частота:
Розповсюдженість спадкового сфероцитозу становить 2-3 на 10000 населення.
Етіологія та патогенез:
В основі захворювання лежить генетичний дефект білка мембрани еритроцита, що призводить до проникнення в еритроцит надлишку іонів натрію та підвищеному накопиченню в ньому води, внаслідок чого утворюються сферичні еритроцити (сфероцити).
Сфероцити, на відміну від нормальних еритроцитів, не мають здатності деформуватися у вузьких ділянках кровотоку, наприклад при переході в синуси селезінки. Це призводить до уповільнення просування еритроцитів в синусах селезінки, відщеплення частини поверхні еритроцита з утворенням мікросфероцитів та поступової їх загибелі. Зруйновані еритроцити поглинаються макрофагами селезінки. Постійний гемоліз еритроцитів в селезінці веде до гіперплазії клітин її пульпи та збільшення органа. В зв’язку з посиленим розпадом еритроцитів в сироватці підвищується вміст білірубіну. Білірубін надходить в кишківник та виводиться з організму з сечею та, головним чином, з випорожненнями у вигляді стеркобіліну. Добове виділення стеркобіліну при спадковому мікросфероцитозі перевищує норму в 10-20 разів.
Наслідком підвищеного виділення білірубіну в жовч є утворення пігментних каменів в жовчному міхурі та протоках.
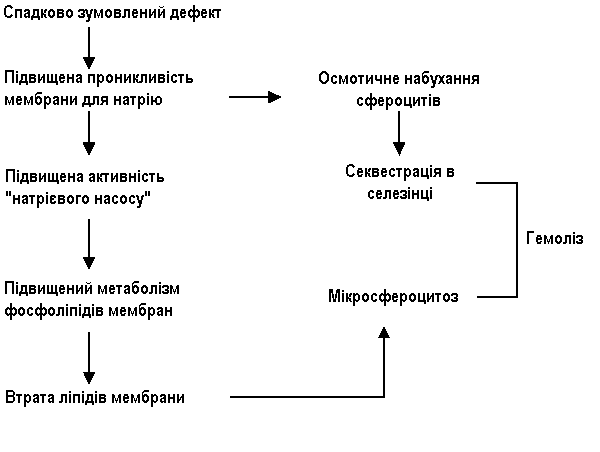 Основні діагностичні критерії:
Основні діагностичні критерії:
Нормохромна анемія, гемолітична жовтяниця, спленомегалія, наявність мікросфероцитозу, симптоми гемолітичної жовтяниці у членів сім’ї.
Клініка:
Клініка залежить від вираженості гемолізу. В більшості випадків перші ознаки виявляються в молодому або зрілому віці. У дітей хвороба виявляється часто при обстеженні з приводу захворювання їх родичів. Скарги поза загостренням можуть бути відсутні. В період загострення відмічаються слабке запаморочення голови, підвищення температури тіла.
Одним з основних клінічних симптомів є жовтяниця, яка довгий час може лишатися єдиною ознакою захворювання. Вираженість жовтяниці залежить з одного боку, від інтенсивності гемолізу, а з другого – від здатності печінки до кон’югації вільного білірубіну з глюкуроновою кислотою. В сечі білірубін не виявляється. Випорожнення інтенсивно забарвлені в темно-коричневий колір внаслідок підвищеного вмісту стеркобіліну.
Кардинальним симтомом спадкового мікросфероцитозу є збільшення селезінки. При тривалому гемолізі спостерігається значна спленомегалія, в зв’язку з чим хворі скаржаться на відчуття тягара в лівій підреберній ділянці.
Печінка при неускладненому захворюванні нормальних розмірів, але іноді у хворих, які довготривало страждають гемолітичною анемією, виявляється її збільшення.
Можуть спостерігатися:
- вторинні аномалії скелету – “баштовий череп”, широка спинка носа, високе піднебіння, вузька альвеолярна дуга, брахі-, син-, або полідактилія; вроджений вивих кульшових суглобів;
- вроджені очні аномалії: мікрофтальм, звужена очна щілина, гетерохромія райдужної оболонки, ексцентричні зіниці, помутніння кришталика та рогівки, астигматизм, часткова кольорова сліпота, вип’ячування очних яблук;
- аномалії вух: прирослі вушні мочки, отосклероз з туговухістю або глухотою;
- вроджені вади серця;
- інфантилізм;
- пігментні аномалії.
Вираженість анемічного синдрому різна. Частково відмічається помірне зниження гемоглобіну. У деяких хворих анемія взагалі відсутня. Найбільш різка анемізація спостерігається в період гемолітичних кризів, які проявляються посиленням симптомів на тлі безперервного гемолізу. При цьому підвищується температура тіла в зв’язку з масовим розпадом еритроцитів, збільшується інтенсивність жовтяниці, з’являються сильні болі в животі, блювота. Гемолітичні кризи виникають, як правило, після інтеркурентних інфекцій, переохолодження, у жінок в зв’язку з вагітністю.
Гемолітичний криз супроводжується анемією, лейкоцитозом, виявленням в крові нормоцитів, гіперплазією еритроїдного паростка кісткового мозку при незначному збільшенні недиференційованих клітин поряд з спленомегалією, що дає привід до помилкової діагностики деяких форм лейкозів.
При диференціальній діагностиці спадкового мікросфероцитозу з іншими гемолітичними анеміями необхідно виключити аутоімунні гемолітичні анемії.
Спадковий мікросфероцитоз може проявлятися в ранньому віці, що певним чином затруднює діагностику. Як правило, рання маніфестація симтомів хвороби свідчить про важкість перебігу. Характерна рання інтенсивна жовтяниця. Поряд із спленомегалією нерідко також збільшена печінка. В крові підвищений рівень як вільного, так і зв’язаного білірубіну. Така клінічна картина примушує проводити диференціальну діагностику з гемолітичною хворобою новонароджених, гепатитом, механічною жовтяницею. Дані анамнезу, проба Кумбса (фіксовані на еритроцитах аутоантитіла), еритроцитометричні показники, осмотична резистентність еритроцитів, ретикулоцитоз мають суттєве значення для правильної діагностики.
Ускладнення:
При спадковому мікросфероцитозі описані арегенераторні кризи. Під час такого кризу з’являються симптоми гіпоплазії кісткового мозку з ураженням еритроїдного паростка. Подібні арегенераторні стани кісткового мозку короткочасні, однак вони зумовлюють важку анемію та можуть спричинити раптову смерть.
Хвороба Мінковського-Шоффара може ускладнюватись утворенням пігментних каменів в жовчному міхурі та жовчних протоках. В зв’язку з цим у хворих спостерігаються симптоми ангіохолецистіту, паренхіматозного гепатиту з підвищенням рівня прямого білірубіну в крові. Описані випадки тромбозу селезінкової вени з наступними шлунковими кровотечами та симптомами гіперспленізму.
Співвідношення статі: Ч1 : Ж1
Тип успадкування:
Аутосомно-домінантний з неповною пенетрантністю.
Вік маніфестації:
Може проявлятися в період новонародженості, в ранньому дитячому віці і на початку шкільного віку.
Лікування:
Єдиним методом лікування хворих спадковим мікросфероцитозом є спленектомія, яка виявляється ефективною в 100% випадків. Після спленектомії у хворих настає практичне одужання, не дивлячись на те, що еритроцити зберігають свої патологічні властивості. Припинення гемолізу після спленектомії пояснюється видаленням основного плацдарму руйнування мікросфероцитів.
Прогноз:
Відносно благоприємний. Більшість хворих доживає до старості. Вірогідність виникнення захворювання у дітей, якщо один з батьків хворіє на мікросфероцитоз, дещо нижча 50%.
Профілактика: бажана генетична консультація.
Спеціальні рекомендації:
Рання діагностика, динамічне спостереження, спленектомія при важкому перебігу захворювання забезпечують практичне одужання хворих.
Номер з каталогу МІМ:
182900 Spherocytosis, Hereditary; HS
Література:
- Козлова С.И., Семанова Е., Демикова Н.С. Наследственные синдромы и медико-генетическое консультирование.- Ленинград, 1987.- С. 206-207.
- Резнік Б.Я., Зубаренко А.В. Практична гематологія дитячого віку.- Київ, 1989.- С. 362-369.
- Fan – Chеn Mong, Kwang – Shin Cho, Kir – Young Kim. A Case of Congenital Microspherocytosis Requiring Early Splenectomy. Yonsei medical journal 1987;28(3):234-42.
- Morton N., Mackinney A.A., Kosower N.S. at al. Genetics of spherocytosis. Am J. Hum. Genet. 1982;14:170-184.
Переглянуто редакційною колегією I.B.I.S.: 27/11/2002
Синдром Штурге-Вебера
(Sturge-Weber Syndrome)
Оксана Миколаївна Масняк
Лікар-генетик
Рівненського обласного медико-генетичного центру
Рівненського обласного клінічного лікувально-діагностичного центру ім. В. Поліщука
Включення:
- Енцефалофаціальний ангіоматоз;
- Енцефалотригемінальний ангіоматоз;
- Менінгеальний капілярний ангіоматоз.
Виключення:
- Нейрофіброматоз;
- Туберозний склероз;
- Синдром Хіппеля-Ліндау.
Основні діагностичні критерії:
Ангіоматоз шкіри, мозку, сітківки.
Класифікація:
Тип І – невус на двох половинах обличчя, лептоменінгеальні ангіоми, може бути глаукома.
Тип ІІ – ангіома обличчя, може бути глаукома.
Тип ІІІ – ізольована лептоменінгеальна ангіома, глаукома відсутня.
В сучасній літературі виділяють:
- трисимптомну форму (нейроокулокутанеальну) у 17,5 % хворих;
- бісимптомну форму (нейрокутанеальну) у 70% хворих;
- моносимптомну форму (кутанеальну) у 12,5% хворих
Ознаки:
Шкірні ангіоми локалізуються, зазвичай, в ділянці першої або всіх трьох гілок трійчастого нерва, переважно з однієї сторони, у вигляді плям від рожевого до пурпурно-червоного кольору. Ураження очей проявляється ангіоматозним переродженням судинної оболонки, частіше на стороні ангіом, можлива вторинна глаукома. Неврологічні зміни – генералізовані судомні напади, геміплегія, геміпарези – зумовлені ангіоматозним ураженням судинної і м’якої мозкових оболонок, а також вторинною атрофією і склерозуванням кори мозку. Можлива розумова відсталість.
Асоційовані вади:
Мікрогірія, макроцефалія, звивистість судин сітківки, гетерохронія райдужки, відшарування сітківки, страбізм, коарктація аорти, макродактилія, капілярні мальформації за межами обличчя.
Дані обстежень:
- Рентгенографія черепа – кальцифікати з характерним їх розміщенням в тім’яно-потиличній ділянці;
- КТ головного мозку – кортикальна атрофія та інтракортикальна кальцифікація типу “трамвайного шляху”; геміатрофія мозку з розширенням бокового шлуночка;
- МРТ головного мозку – дисплазії кори; геміатрофія, гліоз, демієлінізація; контрастне підсилення – ангіоматозна мальформація; розширення шлуночків;
- ангіографія – збільшення глибоких церебральних або колатеральних вен, зменшення або відсутність вен кори, раннє наповнення крупних вен;
- ЕЕГ – основна активність часто аномальна, міжпівкульна асиметрія, реєструються фокальні і мультифокальні спайки і комплекси пік-хвиля в лобних, лобно-скроневих, центральних або тім’яно-потиличних відділах;
- офтальмологічні дані – ангіоматоз повік, звивисті кон’юнктивальні і епісклеральні судинні сплетіння, одностороння гетерохромія райдужки, хоріоїдальна гемангіома, глаукома; ексудативне відшарування сітківки.
Диференційний діагноз:
Нейрофіброматоз, туберозний склероз, синдром Хіппеля-Ліндау, синдром Кліппеля-Треноне-Вебера, синдром Рандю-Ослера, вторинна глаукома.
Етіологія: на даний час вивчена недостатньо.
Частота виникнення: невідома.
Співвідношення між статями:
Невідоме. Ангіоми шкіри спостерігаються у жінок у 3 рази частіше, ніж у чоловіків.
Вік прояву:
Ангіоми шкіри спостерігаються від народження або з’являються в перші місяці життя дитини. Епілептичні напади діагностуються до 1 року в 50%.
Прогноз:
Якщо наявні часті судоми, спостерігається відставання в розумовому розвитку. Нормальний розвиток може спостерігатись у пацієнтів з повільно прогресуючим перебігом захворювання без тяжкого ураження ЦНС. Смертність складає 3,3–14%, найчастіше зумовлена резистентними судомами.
Ускладнення:
Епілептичні напади, відставання в розумовому розвитку.
Лікування:
Симптоматичне. Хірургічне лікування ангіом шкіри і головного мозку проводять при обмежених, ізольованих ураженнях (кріотерапія, лазеротерапія). Іноді при гемангіомі мозку показана променева терапія.
При прогресуванні енцефалотригемінального ангіоматозу хворі помирають, смерть настає під час епістатусу від набряку головного мозку. Інфекційні хвороби і травми можуть ускладнювати перебіг енцефалотригемінального ангіоматозу і призвести до летальних випадків у зв’язку з розвитком субарахноїдально-паренхіматозних крововиливів.
Номер з каталогу МІМ:
185300 Sturge-Weber Syndrome; SWS.
Література:
- Диагностика и лечение наследственных заболеваний нервной системы у детей. Руководство для врачей /Под ред. В.П. Зыкова.– М: ”Триада-Х”, 2007.– С.92–98.
- Козлова С.И., Демикова Н.С., Семанова Е., Блинникова О.Е. Наследственные синдромы и медико-генетическое консультирование.- М.: Практика, 1996.- C.318-319.
- Birth Defects Compendium. D. Bergsma (ed.). Second Edition. New York, 1979:987.
- Gorlin RJ, Cohen MM, Hennekam RCM. Syndromes of the Head and the Neck. 4th edition. Oxford University Press. 2001:456-459.
- Jones KL. Smith’s Recognizable Patterns of Human Malformations. 6th edition. W.B. Sauders Company. Philadelphia. 2002:572.
Переглянуто редакційною колегією I.B.I.S.: 14/10/2010
Пренатальний біохімічний скринінг
(Prenatal Biochemical Screening)
Н.Ф. Маковей
Завідуюча лабораторією імуноферментного аналізу
Хмельницького клінічного пологового будинку
За даними Всесвітньої організації охорони здоров’я 2,5% всіх новонароджених мають різноманітні вади розвитку; 1,5% з них обумовлені дією шкідливих екзогенних факторів під час вагітності, решта мають переважно генетичну природу. Близько 40-50% ранньої смертності новонароджених (перинатальної) та інвалідності з дитинства також обумовлені спадковими факторами.
Вирішальна роль в комплексі заходів по профілактиці та попередженню спадкових і вроджених хвороб належить пренатальній діагностиці (ПД), яка дозволяє попередити народження дітей з важкими, некоригованими вадами розвитку, з соціально значимими смертельними генетичними та хромосомними захворюваннями, і тим самим зменшити генетичний груз популяції.
ПД спадкових і вроджених хвороб – новий розділ медичної генетики, що виник у 80-х роках ХХ ст., на зіткненні клінічних дисциплін (акушерства, гінекології, неонатології) і фундаментальних наук (патофізіології, біохімії, цитогенетики, молекулярної біології, генетики людини). Її основні наукові завдання знаходяться в області біології розвитку (ембріології) людини і включають вивчення особливостей реалізації генетичної інформації зародка, що розвивається в нормі та при патології на молекулярному, клітинному, тканинному рівнях, на рівні організму та розробку на цій основі оптимальних способів профілактики, діагностики і, в майбутньому, лікування спадкових хвороб.
У практичному плані ПД – це комплекс методів, направлених на діагностику морфологічних, структурних, функціональних чи молекулярних порушень розвитку, що проявляються у вигляді ізольованих або множинних вроджених вад, дизрупцій, деформацій, недорозвинення, хромосомних чи моногенних хвороб, у вадах чи дисфункціях життєво важливих систем органів і тканин, які призводять до важких, нерідко смертельних хвороб у постнатальному періоді.
Завдання ПД як одного із розділів медико–генетичної служби включають:
- Надання майбутнім батькам вичерпної інформації про ступінь ризику народження хворої дитини.
- У випадку високого ризику надання інформації про можливість переривання вагітності і наслідках прийнятого батьками рішення – народити хвору дитину чи перервати вагітність.
- Забезпечення оптимального проведення вагітності і ранньої діагностики внутрішньоутробної патології.
- Визначення прогнозу здоров’я майбутнього потомства.
Методи ПД розділяють на непрямі (об’єктом дослідження є вагітна жінка) і прямі (досліджується сам плід). Останні можуть бути інвазивними та неінвазивними.
Основні методи оцінки стану плода в пренатальній діагностиці вроджених спадкових захворювань
- Непрямі методи (обстеження вагітної):
- Акушерсько–гінекологічні;
- Медико–генетичні (генеалогічні, цитогенетичні, молекулярно–біологічні);
- Бактеріологічні, серологічні;
- Біохімічні скринінгові тести на альфа-фетопротеїн, естріол, хоріонічний гонадотропін та ін.)
- Прямі методи:
- Неінвазивні (ультразвукове сканування, електрокардіографія, рентгенографія та ін.);
- Інвазивні:
- Хоріонбіопсія:
- трансвагінальна;
- трансабдомінальна;
- Плацентоцентез;
- Кордоцентез;
- Фетоскопія;
- Біопсія тканин плода (печінка, селезінка, шкіра, м’язи та ін.)
У даному огляді детальніше зупинимося на біохімічному скринінгу, метою якого є виявлення осіб з високим ступенем ризику виникнення ряду порушень до появи у них клінічних симптомів. Пренатальна біохімічна діагностика використовує визначення концентрації різноманітних сполук (білкових і стероїдних гормонів, амінокислот та ін.), що виробляються організмом матері, плацентою і плодом, для виявлення і моніторингу лікування патологічних станів вагітності, включаючи порушення розвитку плода. При впровадженні біохімічного скринінгу вагітних до уваги приймаються наступні фактори:
- захворювання, виявлене за допомогою скринінгу, чітко розпізнається;
- проведення скринінгу обходиться дешевше, аніж лікування захворювань;
- аналіз, що використовується при скринінгу, повинен бути високоспецифічним (тобто рідко давати хибнопозитивні чи хибнонегативні результати) та високочутливим (виявляти високий процент прихованої форми захворювання).
Слід мати на увазі, що при використанні усіх скринінгових систем, що базуються на визначенні біохімічних маркерів у допологовому періоді, можливе отримання хибнопозитивних результатів, тобто отримання патологічних значень при нормальному розвитку плода. В оптимально налагоджених системах кількість вроджених вад розвитку плода складає 1:20 від кількості позитивних результатів. Водночас, певний відсоток захворювань залишається невиявленим. Кількість хибнонегативних результатів складає 10-20%.
Біохімічні дослідження маркерних сироваткових білків крові вагітних, як і ультразвукове дослідження зараз розглядають як обов’язкові скринінгові методи допологової діагностики, направлені на виявлення жінок груп високого ризику народження дітей із хромосомними аномаліями і вадами розвитку.
До маркерних білків крові матері відносять альфа-фетопротеїн (АФП), хоріонічний гонадотропін (ХГ), вільний некон’югований естріол (НЕ), інгібін-А, асоційований з вагітністю білок плазми А (РАРР-А) та деякі інші. Всі ці білки є ембріонспецифічними, тобто продукуються клітинами самого плода, поступають у кровотік матері. Їх концентрація в сироватці крові зменшується залежно від терміну вагітності та від стану плода. З їх допомогою пренатальний біохімічний скринінг ідентифікує вагітних жінок їз підвищеним ризиком розвитку у плода синдрому Дауна (трисомія 21 хромосоми), синдрому Едвардса (трисомія 18 хромосоми), або вад невральної трубки (ВНТ). Для інших хромосомних хвороб, наприклад, синдрому Тернера, скринінг неспецифічний.
Значення серологічних маркерів можуть мати різноманітні варіації в різних лабораторіях. Для того, щоб коректно порівнювати результати, відхилення рівня маркера від норми виражають, як правило, через кратність медіани. Медіана є середньою серед упорядкованих по зростанню значень рівня маркера при нормальній вагітності того ж терміну гестації – MoM (Multiples of median), наприклад, рівень АФП = 2,5 МоМ, отже, вміст білка в сироватці крові даної вагітної в 2,5 рази вище, ніж медіана (норма) для цього терміну вагітності. Вірогідність народження хворої дитини при певному значенні МоМ розраховано статистично на великій кількості випадків (мал. 1).
Хоріонічний гонадотропін
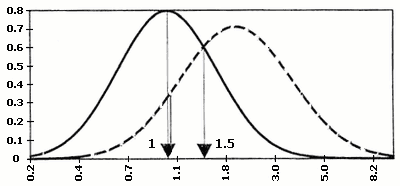
АФП
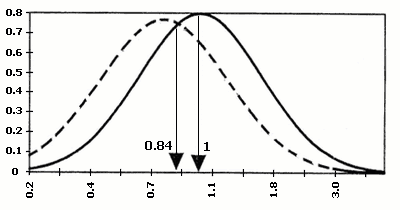
Некон‘югований естріол
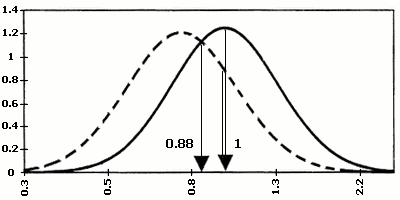
Мал.1. Діаграми розподілу по концентраціях індивідуальних маркерів ХГ, АФП, НЕ
в сироватці крові матері при вагітності плодом з синдромом Дауна та в нормі.
АФП – основний компонент фетальної сироватки на ранніх термінах вагітності – глікопротеїн масою 68 кΔа, синтезується жовточним мішком плода і в 2 триместрі – печінкою плода, а також у незначній кількості він утворюється клітинами кишкового тракту, нирок, плаценти. Білок екскретується в амніотичну рідину з сечею плода, проникає в кров матері через плаценту і всмоктується через плідні оболонки. За своїми фізико-хімічними властивостями АФП близький до сироваткового альбуміну. Функції АФП остаточно не з’ясовані, однак головними із них є: підтримання онкотичного тиску, імуномодуляція; зв’язування материнських естрогенів, участь в органогенезі печінки; властивості транспортного білка. Наприкінці 1 триместру вагітності жовточний мішок атрофується, і основним місцем синтезу АФП стає печінка плоду, звідки цей білок потрапляє у кровообіг плода, утилізується нирками та екскретується із сечею в амніотичну рідину. Далі АФП разом із амніотичною рідиною заковтується плодом і перетравлюється. У кров матері АФП проникає шляхом дифузії, або активного транспорту через плідні оболонки, плаценту, судини пуповини. Вміст АФП у сироватці плода, амніотичній рідині та материнській сироватці визначається гестаційним терміном. Максимальна концентрація АФП у фетальній сироватці спостерігається між 10 та 13 тижнями вагітності, поступово знижуючись до народження. Концентрація АФП в навколоплідних водах строго корелює із його вмістом у крові плода.
Динаміка змін концентрації АФП в материнській сироватці відрізняється: вміст АФП у сироватці матері збільшується з кінця першого триместру (білок виявляється в крові матері, починаючи з 5-6 тижнів), сягаючи максимальних значень на 32-33 тижнях вагітності, далі поступово знижується до пологів. Враховуючи той факт, що фізіологічне збільшення концентрації АФП в крові матері з 13 до 33 тижнів гестації проходить на фоні зниження його вмісту в крові плода, можна припустити, що він безпосередньо відображає проникність плаценти. Особливо це стосується другої половини гестації. На початку ІІ триместру вміст АФП у сироватці вагітної в основному залежить від його дифузії із амніотичної рідини через плідні оболонки.
Оптимальним терміном для дослідження материнської сироватки вважається 15–20 тижні гестації. До 15 тижнів концентрація АФП недостатньо надійна, як маркер дефектів невральної трубки, а понад 20 тижнів рівень АФП характеризує функціональну ступінь зрілості плода.
Вади невральної трубки призводять до виливання фетальної рідини в амніотичну порожнину, внаслідок чого різко збільшується концентрація альфафетопротеїну в крові матері, тому підвищення його рівня в материнській сироватці в 2 триместрі вагітності з високим ступенем вірогідності вказує на наявність у плода вроджених вад невральної трубки. Суттєве підвищення рівня АФП в крові матері спостерігається також при інших патологічних станах (табл. 2). В той же час у 30% випадків хромосомних порушень у плода (синдром Дауна) в діагностичному терміні (15-20 тиждень вагітності) виявляється зниженим (табл. 2).
Таблиця 2. Причини підвищення і зниження
рівня материнського сироваткового АФП під час вагітності.
| Аненцефалія Рахисшис (розщелина хребта) Ембріональна кила Гастрошизис (дефект передньої черевної стінки) Торакоабдомінальний дефект Вроджена відсутність нирок Тератома Гідроцефалія Множинні вади розвитку Гігрома шиї Маловіддя Синдром Меккеля Енцефалоцеле |
Синдром Дауна та інші хромосомні аберації |
| Позаматкова вагітність Прееклампсія Порушення форми плаценти Вірусний гепатит Первинна карцинома печінки Злоякісні пухлини шлунково–кишкового тракту Пухлини зародкового походження |
Цукровий діабет 1-го типу Хоріон аденома Хоріокарцинома |
| Загибель плода Дисфункція плаценти Резус-несумісність |
|
Стан матері та плода, що впливає на материнський рівень АФП
(вірогідність патології плода 90%, за відсутності акушерських причин для підвищення АФП) |
аненцефалія, відкрита спинно-мозкова кила, енцефалоцеле |
відкрите пошкодження у плода |
омфалоцеле, гастрошизис |
||
при аутолізі плода із вивільненням АФП в амніотичну рідину |
||
із сечею плода, підвищення його концентрації в крові плода |
||
АФП, який заковтується плодом разом із амніотичною рідиною |
||
супроводжується шийною гігромою (синдром Тернера, трисомія 13) |
||
(у поєднанні із підвищеним рівнем ХГ) |
||
та самовільного викидня |
АФП в кровотік матері при посиленому скороченні мускулатури матки |
|
ХГ – глікопротеїн, який синтезується трофобластами плаценти, має молекулярну масу 40кΔа, складається з двох субодиниць α і β (α-cубодиниця спільна із гормонами гіпофізу ТТГ, ФСГ, ЛГ, а β-субодиниця визначає специфічність). ХГ виявляється в сироватці крові жінок, починаючи з 10-12 дня після запліднення, тобто на 3-5 день після імплантації. Його концентрація швидко зростає і досягає максимуму на 8-10 тижні вагітності. На перших тижнях вагітності концентрація ХГ подвоюється кожні два дні. Із плаценти ХГ потрапляє переважно в кров матері і там його концентрація у 10-20 разів перевищує відповідну концентрацію у плода. Період напіврозпаду ХГ становить 27 годин, тому повторне його визначення з інтервалом у декілька днів відображатиме істинну зміну його секреції. Біологічна роль ХГ характеризується:
- лютеотрофний ефект: підтримка синтезу прогестерону жовтим тілом яйників у першій третині вагітності та синтез естрогенів плацентою у другій половині вагітності, тобто забезпечення гормонального фону для нормального розвитку вагітності;
- регуляція ендокринної системи плода: стимуляція синтезу стероїдних гормонів у його наднирниках, яйниках чи яєчках;
- пригнічення продукції ФСГ для запобігання появі звичайних менструальних змін в ендометрії;
- запобігання імунологічному відторгненню плода організмом матері.
Рівень ХГ при синдромі Дауна у плода, як правило, підвищується, а при трисомії хромосоми 18 (синдром Едвардса) – знижується. Крім того, зниження рівня ХГ відбувається при вагітності, що не розвивається, та при загрозі її переривання. Зниження відбувається паралельно з прогестероном, що обумовлено порушенням гормональної активності жовтого тіла вагітності. Поряд з ХГ часто використовують також визначення β-субодиниці ХГ, яка має те ж діагностичне значення.
НЕ – фетоплацентарний стероїдний гормон масою 288,4 Δа, один з трьох головних естрогенів, разом з естрадіолом та естроном. Синтезується фетальними печінкою, наднирниками і плацентою, тому є ідеальним показником функції фетоплацентарної системи. На першій стадії синтезу, яка проходить в ембріоні, холестерин, що утворюється de novo, або поступає з крові матері, перетворюється в прегнеколон, який сульфатується корою наднирників плода в дегідроепіандростерон–сульфат. Гідроксилювання цієї сполуки в печінці по 16 α-положенню і відщеплення сульфату сульфатазами плаценти призводить до утворення естріолу. В материнській крові лише 9% естріолу циркулює у вільному стані, більша його кількість знаходиться у вигляді глюкуроніду і сульфату. Під час фізіологічної вагітності рівень некон’югованого естріолу поступово зростає до 40 тижня, безпосередньо характеризуючи функціональний стан плаценти і плода. Недостатній синтез НЕ викликає загрозу переривання вагітності в першій половині і загрозу передчасних пологів в другій. Зниження рівня НЕ спостерігається не лише при синдромі Дауна, а і при вірилізуючій формі вродженої надниркової гіперплазії, при дефіциті плацентарної сульфатази чи 16-α-гідроксилази плоду, аненцефалії, внутрішньоутробній інфекції. Зниження концентрації в плазмі крові більше ніж 35% (при нормі від 4 нмоль/л в 15 тижнів вагітності до 40 нмоль/л при пологах), вказує на гостру недостатність функціонування фетоплацентарного комплексу і є ознакою загрози для плоду. Показниками для дослідження є вагітність з високим ризиком, включаючи цукровий діабет, переношування, прееклампсію, затримку росту плоду, резус–конфлікт, внутрішньоутробну загибель плоду, анемію, порушення харчування, пієлонефрит, захворювання кишківника і гемоглобінопатію, гіпоплазію наднирників плоду, аненцефальний плід.
Інгібін–А – гетеродимерний гормон білкової природи, що гальмує секрецію фолікулостимулюючого гормону (ФСГ) гіпофізом. Інгібін–А складається з α та βА – субодиниць; інгібін–Б – з α- та βВ–субодиниць. Тільки димерні форми володіють біологічною активністю. В першому триместрі вагітності, крім яєчників, синтезуються плодом, плацентою та плідними оболонками. Протягом вагітності підвищується до 10 тижня, потім знижується і залишається стабільним з 15-25 тижня, потім знову зростає до пікових значень перед пологами. Його визначення має діагностичне значення для пренатального скринінгу синдрому Дауна.
Асоційований з вагітністю білок плазми А (РАРР-А) в сироватці крові знаходиться в комплексі головного основного протеїну еозинофілів (proeosinophil major basic protein, proMBP), при чому дві його субодиниці пов’язані з двома ланцюгами останнього дисульфідними містками. Амінокислотна послідовність РАРР-А на значному проміжку унікальна. Лише в С-кінцевому відділі є ділянки, гомологічні білкам системи комплемента і селектинам. Крім того, три фрагменти його ланцюга виявляють подібність з факторами, що регулюють ранню тканинну диференціацію. Білок включено до сімейства α-2-мікроглобулінів людини. РАРР–А є IGF–II-залежною протеазою, що розщеплює IGFВР–4-білок (IGF–II – інсуліноподібний фактор росту ІІ, IGFВР–4-білок, що зв’язує інсуліноподібний фактор росту), та синтезується фібробластами.
РАРР-А пригнічує проліферативну активність лейкоцитів, тому він входить в групу білків–імуносупресорів, до яких відноситься ХГ, трофобластичний β-1-глобулін, АФП і асоційований з вагітністю α-2–глікопротеїн. Ці білки in vivo забезпечують пригнічення імунологічної реактивності материнського організму стосовно зародку, що розвивається.
При вагітності рівень РАРР-А різко збільшується за рахунок синтезу плацентою (протягом перших 8 тижнів вагітності концентрація РАРР-А в сироватці крові подвоюється кожні 4,9 діб, а до 10 тижня збільшується приблизно у 100 разів, градієнтно збільшуючись протягом усієї вагітності, і у багатьох жінок напередодні пологів перевищуючи 100 мкг/мл).
Білок вважається кращим біохімічним маркером 1 триместру для діагностики синдрому Дауна. Має чутливість 42% при 5% хибно позитивних результатів. Молекула білка характеризується високим ступенем глікозилювання, який, імовірно, вищий при вагітності плодом з трисомією 21.
РАРР-А має більшу стійкість до тіолових протеїназ, ніж ХГ, який змінює свій рівень внаслідок протеолітичного розщеплення гормону. Можлива об’єктивна оцінка концентрації РАРР-А при зберіганні (пересиланні поштою) сироватки на паперових фільтрах, що, безумовно, згодом полегшить масовий скринінг вагітних.
Білок–1, зв’язуючий імуноподібні фактори росту (IGFВР–1) – фосфопротеїн з молекулярною масою 25,3 кΔа, зв’язує імуноподібні фактори росту І та ІІ. Це один з 6 структуроподібних білків, що зв’язують та моделюють активність інсуліноподібних факторів росту. Синтезується, насамперед в печінці та децидуальній оболонці, в невеликих кількостях може синтезуватися в клітинах гракулози в лютеальну стадію менструального циклу. Більша концентрація IGFВР–1 була ідентифікована в крові вагітних при нирковій недостатності, недоїданні, при інсулін-залежному цукровому діабеті, цирозі та карциномі печінки. Низькі значення IGFВР–1 знайдено у огрядних пацієнтів і при станах, пов’язаних з гіперінсулінемією. IGFВР–1 відносно вище у випадках внутрішньоутробної затримки росту і нижче у випадках вагітності великим плодом. Сироваткові рівні IGFВР–1 змінюються значно в залежності від характеру харчування.
Діагностичне значення визначення IGFВР–1: Високий рівень в амніотичній рідині пов’язаний з порушенням пренатального росту в 2-3 триместрі вагітності. Він є показником до гістологічного дослідження плаценти на 24-25 тижні вагітності. Збільшується в середньому до 20 раз в загинальному секреті в 75% випадків при розриві фетальної мембрани. Трисомія 18 (синдром Едвардса) діагностується високими значеннями IGFВР–1 в сполученні з низькими концентраціями IGFВР–2 в першому триместрі. Залежно від потужності лабораторій для скринінгу використовують ті чи інші білкові маркери.
Скринінг І триместру
Комбінований тест (виконується на 10-13 тижні вагітності) включає визначення вільної субодиниці β-ХГ людини, РАРР-А і NT (комірцевий простір плоду на УЗД). У першому триместрі РАРР-А і NT, як правило, збільшуються з розвитком вагітності. При розвитку у плода синдрому Дауна рівень РАРР-А в материнській крові, в середньому, в два рази менший. NT збільшений також приблизно в 2 рази порівняно з нормою. В цьому тесті можливо визначити 85% всіх плодів з патологією (близько 5% жінок з групи ризику), проте неможливо визначити дефекти відкриття невральної трубки (немає АФП, оскільки визначення АФП в першому триместрі ще немає діагностичної цінності для даного дефекту)
Скринінг ІI триместру
Квадро–тест (виконується на 15-22 тижні вагітності). Найбільш розповсюджений і загальноприйнятий на сьогодні тест пренатального скринінгу синдрому Дауна і трисомії 18.
Квадро–тест – метод скринінгу з використанням вимірювання чотирьох маркерів в материнській сироватці. Це АФП, ХГ, НЕ та інгібін–А. Ці серологічні маркери і вік жінки використовуються разом для оцінки ризику вагітності плодом з синдромом Дауна. В нормі в другому триместрі рівні АФП і НЕ ростуть (15% і 24% відповідно в тиждень), рівень ХГ зменшується, рівень інгібіну–А повільно знижується перед 17 тижнем і так само повільно зростає після 17 тижня. При вагітності плодом з синдромом Дауна АФП і рівні НЕ, в середньому, нижче Мом на 75%. Якщо використовується скорочений варіант скринінгу, то чутливість виявлення плода із синдромом Дауна набагато нижча:
- При одночасному використанні двох маркерів (АФП і β-ХГ) в сироватці жінок вірогідність діагностики синдрому Дауна у плода складає 59%;
- При визначенні трьох маркерів (так званого “потрійного” тесту: АФП, β-ХГ і НЕ) – 69%;
- При визначенні чотирьох біохімічних маркерів (АФП, β-ХГ, НЕ та інгібіну–А) і з включенням у розрахунки терміну вагітності і віку матері – 79% при 5% рівні хибно позитивних результатів.
Скринінг І та ІІ триместрів
Інтегральний тест (двохетапний пренатальний скринінг синдрому Дауна і трисомії 18) – найбільш ефективний метод скринінгу. Тест виконується в дві стадії:
- Перша стадія – ідеально проводиться в 12 тижнів (проте може бути будь–який проміжок часу між 10 і 13 тижнем), коли береться проба крові на аналіз РАРР-А і паралельно проводиться ультразвукове дослідження NT (комірцевий простір, розвиток кісток носа);
- Друга стадія – включає забір другої проби крові приблизно через 3-4 тижні після забору першого зразка на АФП, ХГ, НЕ та інгібін–А. Оптимально кров береться на 16 тижні вагітності, але можливо зробити аналіз до 22 тижня.
Ці біохімічні маркери + NT + вік жінки використовують для оцінки ступеня ризику.
Інтегральний тест, хоч і заснований на інформації, отриманій від двох зразків для аналізу, і в першому, і в другому триместрі, є новим підходом в пренатальній діагностиці синдрому Дауна. Якщо розрахувати сумісний ризик по результатах інтегрального тесту, то ефективність виявлення синдрому Дауна у плода може досягати 84-90% з 2% хибно позитивних результатів. Це щонайменше на 20% покращує традиційний потрійний тест.
Слід мати на увазі, що найбільш розповсюдженою причиною хибнопозитивних результатів скринінгу буває невірний підрахунок терміну вагітності, тому при невідповідності рівня білка нормі необхідно, в першу чергу, ще раз уточнити термін вагітності за допомогою УЗД.
Нормальними значеннями в діагностичні терміни вважаються рівні білків від 0,5 до 2,5 Мом.
Використання незалежних маркерів (тобто слабко коригованих один з одним) в комбінації збільшує чутливість і специфічність методу і дозволяє прораховувати індивідуальний ризик. Отримані за межею прийнятої норми результати для даних показників вважаються позитивними (див. Табл.3). Вірогідність народження хворої дитини при певному значенні Мом розрахована статистично на великій кількості прикладів. У конкретної пацієнтки вірогідності, отримані для кожного маркера, підсумовуються. Показником для застосування інвазивної пренатальної діагностики з метою каріотипування плода зазвичай рахується ризик народження хворої дитини вище, ніж 1:250 (від 1:190 до 1:400 в різних країнах).
Табл. 3. Рівень в Мом для різних патологій
| Вроджена патологія | ||||||
|---|---|---|---|---|---|---|
| Синдром Дауна | ||||||
| Дефекти закриття нервової трубки | ||||||
| Синдром Едвардса |
Результатом біохімічного скринінгу є відбір вагітних групи ризику народження дітей з вадами невральної трубки (ВНТ) і з хромосомними хворобами плода.Таким вагітним необхідна інвазивна пренатальна діагностика. У випадку високого ризику ВНТ у плода показано ультразвукове дослідження з наступною інвазивною процедурою – амніоцентезом і аналізом амніотичної рідини на вміст АФП і наявність специфічної ізоформи ацетилхолінестерази, характерної для плодів з ВНТ. При відхиленнях маркерних білків, які свідчать про можливість хромосомних хвороб у плода, проводиться інвазивна процедура з метою каріотипування плода. При проведенні скринінгових досліджень вагітну слід інформувати про можливість отримання кваліфікованої інтерпретації результатів лабораторного аналізу і забезпечити при необхідності її направлення на подальшу діагностику.
При масовому скринінгу число вагітних, яким показана інвазивна процедура, як правило, складає близько 5% від загальної кількості обстежених. Важливо розуміти, що позитивні результати (екстремальне відхилення рівня білків від медіани) скринінгу – лише сигнал до більш глибокого обстеження цієї пацієнтки, позаяк хромосомна патологія буде знайдена приблизно у однієї з 50 вагітних цієї групи, а ВНТ – у однієї з 400 обстежених.
Вроджена патологія, включаючи структурні вади, хромосомні аномалії та метаболічні порушення, є однією з найбільш розповсюджених причин перинатальної смертності. На жаль, первинна профілактика охоплює лише малу частину вроджених аномалій. Третинна профілактика, тобто консервативне лікування чи хірургічна корекція, приносить певні результати, проте нерідко пацієнти після складного і тривалого лікування залишаються інвалідами і мають багато проблем. Тому одним з головних резервів зниження рівня перинатальної захворюваності та смертності є вторинна профілактика та пренатальна діагностика вродженої і спадкової патології.
Література:
- Грініо Л.П., Агафонов Б.В. Міопатії.- Москва, 1997. – C. 287.
- Баєв А.А. Вводные замечания //Итоги науки и техники: Геном человека: Т.Г. – Москва: ВИНИТИ, 1994. – C.3-8.
- Карпищенко А.И. Медицинская лабораторная диагностика (программы и алгоритмы). – Санкт–Петербург: Интермедика, 1997. – С. 180–184.
- Компания «Immunotech». Скрининг врожденных пороков развития. – Прага, 1997.
- Гордієнко І.Ю. Пренатальна діагностика вродженої і спадкової патології.
- Mc.Kusick VA. Mendelian inheritance in man: a catalog of human genes and genetic disorders. 11 ed. Baltimore. Johns Hopkins University Press. 1994.
Переглянуто редакційною колегією I.B.I.S.: 12/08/2004
|
|
|
|
|
|




