
Uncategorized
Дослідження ворсинок хоріону
(Chorionic Villus Sampling)
І.Є. Харитонова
Координатор Українського Альянсу
із запобігання вродженим вадам розвитку
Дослідження ворсинок хоріону (ДВХ) – це метод, що застосовується у пренатальній діагностиці (допологове обстеження) і може підтвердити чи виключити хромосомні та деякі спадкові захворювання. Звичайно, це дослідження проводиться між 10 та 12 тижнями вагітності, рахуючи від дати останньої менструації жінки.
Кому рекомендують ДВХ?
Це дослідження не пропонують всім без винятку вагітним жінкам, тому що воно може спровокувати викидень, а можливо і інші ускладнення.
ДВХ проводиться в тих випадках, коли є підвищений ризик виникнення хромосомної патології та спадкових хвороб, і батьки хочуть з’ясувати це під час вагітності якнайраніше. За допомогою іншого пренатального дослідження, що має назву амніоцентез, також можна виявити ті самі генетичні відхилення, але воно проводиться трохи пізніше, звичайно між 15 та 18 тижнями, рахуючи від останньої менструації жінки. Однак останнім часом почали проводити амніоцентез на більш ранніх етапах вагітності (так само, як і дослідження ворсинок хоріону, – у 12 тижнів).
Дослідження ворсинок хоріону пропонується у таких випадках:
- Вік матері. Ризик народження дитини з певними хромосомними захворюваннями підвищується з віком жінки. Якщо жінці на час вагітності 35 років чи більше, лікарі пропонують їй проходження пренатальних досліджень (амніоцентезу чи біопсії ворсинок хоріона) на наявність хромосомної патології. Найбільш поширеною є хвороба Дауна, що є комбінацією розумових і фізичних розладів, спричинених наявністю додаткової хромосоми. Хвороба Дауна трапляється приблизно у одному випадку на 1250 дітей, якщо жінка народила у 20-річному віці. Кількість випадків зростає до одного на кожні 385, коли жінка народжує у 35 років та одного на 106, коли вік матері становить понад 40 років.
- Попередня дитина чи вагітність з хромосомними розладами чи спадковими хворобами. Якщо у подружжя вже є дитина (чи була вагітність) з хромосомними розладами чи спадковими хворобами, вони повинні пройти пренатальне обстеження під час наступних вагітностей.
- Випадки спадкових захворювань у родині. Подружнім парам пропонується пренатальне обстеження, якщо у них вже є нормальна дитина, і, хоча вона не має вроджених аномалій, але в родині є випадки спадкових хвороб чи хромосомних захворювань, існує підвищений ризик успадкувати генетичний розлад. Пренатальне обстеження пропонується тільки у тих випадках, коли генетичні розлади можуть бути виявлені пренатально. Пренатальна діагностика можлива для всіх звичайних хромосомних хвороб, але не для всіх спадкових.
Дослідження ворсинок хоріону небажані, якщо є:
- Вагінальна кровотеча і кров’янисті виділення під час вагітності.
- Якщо в родині вже були випадки вад невральної трубки (вроджені вади спинного чи головного мозку, включно із spina bifida та аненцефалією). Дослідження ворсинок хоріону не встановить цих захворювань. Однак їх можна діагностувати за допомогою амніоцентезу та/або інших досліджень, наприклад, біохімічних.
- Лікар чи лабораторія не мають досвіду в проведенні методик, що використовуються для ДВХ. Можна звернутися до інших медичних закладів. Як альтернатива може бути запропонований амніоцентез.
Що дає ДВХ?
Для проведення дослідження необхідний маленький шматочок ворсинок хоріона, що є тканиною, яка прикріплює плідний мішок до стінки матки. В лабораторії проведуть аналіз клітин ворсинок, які в нормі є такими ж самими за генетичною та біохімічною побудовою як і ембріон. Результати остаточно готові за 10 днів, хоча попередні результати можуть бути готові і раніше.
Яким чином отримуються зразки матеріалу?
Перш за все, піхва та шийка матки ретельно обробляються антисептиком. Потім, під контролем ультразвуку, лікар вводить тонку трубочку-катетер через піхву та шийку матки (трансцервікальний доступ) до місця, де знаходяться ворсинки, і обережно забирає через шприц маленький зразок ворсинок. Існують два доступи для забору ворсинок хоріона. Вони однакові за рівнем безпеки, якщо тільки у жінки немає відхилення матки назад, що може спричинити викидень у тому разі, коли буде надана перевага трансцервікальному доступові. Тому у випадках з такою будовою матки має бути застосований трансабдомінальний доступ (через черевну стінку). Якщо розташування плаценти перешкоджає проведенню цієї процедури, як альтернатива, має бути проведений амніоцентез.
Після взяття зразка матеріалу, поки жінка ще знаходиться у кабінеті лікаря, за допомогою ультразвуку перевіряються серцеві поштовхи плода. Лікарі рекомендують відпочити кілька годин після проведенню ДВХ. Згідно американських досліджень кожна п’ята жінка відчуває після цього дослідження підвищення тонусу матки; кожна третя має невелику кровотечу чи кров’янисті виділення, що звичайно минають за кілька днів.
Чи є ДВХ небезпечним?
В Сполучених Штатах це дослідження проводиться з 1983 року, а в цілому в світі його пройшли більше як 150 тисяч жінок. Статистичні дослідження показують, що ДВХ провокує викидень трохи частіше ніж амніоцентез. Рівень викиднів, що трапляються після амніоцентезу становить менше одного на 200 обстежень. Для біопсії ворсинок хоріона він становить один-два на кожні 100 для жінок з нормальною будовою матки. Цей ризик може зростати до п’яти на 100 викиднів для жінок з відхиленням матки назад, у яких забір матеріалу для дослідження був через трансцервікальний доступ.
Вивчення причин втрати вагітності, що пов’язані з ДВХ показали, що дуже важливим є майстерність лікаря, який виконує цю процедуру. До того ж, найкращі результати мали ті заклади, що практикували як трансцервікальний, так і трансабдомінальний шляхи забору матеріалу, оскільки є можливість вибору безпечнішого доступу для конкретного пацієнта.
В 1991 році з’явилось декілька публікацій про причетність ДВХ до відсутності чи зменшення кількості пальців на руках і ногах у новонароджених, матері яких проходити це дослідження. Ці вади відмічалися переважно тоді, коли ДВХ проводилось раніше 10 тижнів після останнього менструального періода. Зараз повсюди ДВХ роблять тільки у 10 тижнів, або ще пізніше, а жінки мають бути проінформованими про можливість підвищеного ризику вкорочення кінцівок та інших вад розвитку у плода, пов’язаними з технікою проведення процедури.
Чи є добрі результати ДВХ запорукою народження здорової дитини?
Близько 95% жінок з групи ризику, які пройшли пренатальну діагностику, отримують запевнення в тому, що їх ще ненароджена дитина не матиме розладів, на які вони проходили обстеження. Однак, жоден з пренатальних тестів не може гарантувати народження здорової дитини, оскільки тільки певні вади розвитку можна виключити пренатально. Три чи чотири дитини на кожні 100 новонароджених все ж таки мають вроджені вади.
Результати ДВХ у виявленні певних хромосомних вроджених вад і особливих генетичних проблем є дуже точними (більше за 99%). Однак, схоже, що ДВХ дещо більше за амніоцентез дає непевні результати, лишаючи без відповіді запитання, на які часто можна відповісти після проведення амніоцентезу.
Чи може лікар вилікувати вроджені вади за допомогою ДВХ?
Існує багато вроджених вад розвитку та спадкових хвороб, які, на жаль, є невиліковними. Однак, рання пренатальна діагностика за допомогою ДВХ допомагає лікарю у корекції деяких вроджених вад під час вирішальних моментів розвитку плода.
Наприклад, вроджена гіперплазія наднирників, спадкова вада, що виникає внаслідок нестачі важливого фермента, в результаті чого у плода жіночої статі до 16 тижнів вагітності розвиваються аномальні статеві органи. Лікування плода гормонами у критичні терміни розвитку між 10 та 16 тижнями вагітності може запобігти необхідності оперуватись після народження. Зараз, коли відомо, що є ризик гиперплазії наднирників, пренатальна діагностика за допомогою ДВХ може відповісти лікарю на питання чи потрібно проводити подібне лікування.
Якщо для покращення стану плода пренатальне лікування неможливе, а діагноз поставлений до народження, в цих випадках батьки можуть морально підготуватися і планувати разом з лікарем найбезпечніший вибір, місце і метод проведення пологів, а також спеціалістів, що можуть знадобитися відразу після народження.
Чи потрібно мені робити ДВХ?
Проходити чи не проходити пренатальну діагностику, відповідь на це питання отримують під час особистих обговорень між майбутніми батьками та медичними працівниками. Медичні генетики, лікарі, кваліфіковані дорадники з релігійних питань мають надавати батькам допомогу у прийнятті рішення про проходження пренатальної діагностики, а також про інші питання дітонародження. ДВХ проводиться на ранніх етапах вагітності, і рішення має бути дуже зваженим і уважно спланованим, оскільки є ризик щодо плоду. Це тільки одна з причин, чому так важливо щоб жінка отримувала ранню допологову допомогу, починаючи з відвідин лікаря – гінеколога навіть ще до зачаття.
Подружнім парам, яким треба вибирати між ДВХ та амніоцентезом, треба зважати на багато чинників, а саме можливість вибору техніки виконання, стан здоров’я жінки та переваги методу, і що потрібно встановити за проведеними дослідженнями. За одним важливим винятком (вади нервової трубки), ДВХ чи амніоцентез визначають однакові генетичні розлади.
Чи є шляхи зниження ризику вроджених вад?
Є декілька важливих речей, що можуть робити всі жінки, щоб підвищити шанси на здорову вагітність та народження здорової дитини.
- Плануйте вагітність ще до зачаття після консультації з лікарем.
- Вживайте 0,4 мкг фолієвої кислоти (можна у складі полівітамінів) щодня що настання вагітності і протягом перших трьох місяців вагітності – для запобігання вадам нервової трубки.
- Отримуйте ранню та регулярну пренатальну медичну допомогу.
- Споживайте різноманітну їжу.
- До зачаття відрегулюйте свою вагу.
- Не вживайте напоїв, що містять алкоголь під час всієї вагітності.
- Не вживайте наркотиків; якщо є потреба у ліках, повідомте лікаря, що ви вагітні і проконсультуйтесь з ним.
- Не паліть під час вагітності.
Переглянуто редакційною колегією I.B.I.S.: 1/11/2000
Дивіться також:
Скринінг материнської крові
(Maternal Blood Screening)
Тетяна Сороцька
Спеціаліст з інформаційного забезпечення
Рівненського обласного клінічного лікувально-діагностичного центру
У наш час більшість лікарів пропонують своїм вагітним пацієнткам у рутинному порядку (без спеціальних медичних показів) зробити аналіз крові, який вказує, при наявності, на вищий від середнього ризик деяких серйозних вроджених вад, таких як spina bifida (незарощений хребет) та синдром Дауна (хромосомне порушення). За допомогою цього виду аналізу крові, який називається потрійним, можна отримати дуже корисну інформацію про розвиток плоду. При цьому важливо, щоб кожна вагітна жінка пам’ятала, що у більшості випадків позитивний результат аналізу ще не означає, що є проблеми із плодом, і в цьому можна переконатись завдяки подальшим обстеженням.
Які показники вимірюють при потрійному аналізі крові?
На даний момент цей аналіз служить для визначення рівня трьох речовин в материнській крові.
На початку 80-х років, коли тільки почали робити аналіз материнської крові, визначали тільки рівень альфа-фетопротеїну (АФП), речовини, яку виробляє печінка плоду. Якась частина цього протеїну виділяється в амніотичну рідину, що оточує плід. АПФ в невеликій кількості просочується в кров матері, і його концентрація там поступово зростає аж до пізнього періоду вагітності.
Сьогодні за допомогою цього аналізу, поряд з рівнем альфафетопротеїну в сироватці материнської крові (МСАФП), виявляють рівень двох гормонів – естріолу та хоріонічного людського гонадотропіну (ХЛГ). Тому цей аналіз і називається потрійним, хоча інколи його ще називають АФП3 або покращений АФП.
Щонайменше дві з великих лабораторій у Сполучених Штатах почали вимірювати рівень ще одного компоненту материнської крові, який називається інгібін А. Дослідники продовжують вивчати інші речовини в материнській крові, які згодом, можливо, теж будуть вимірювати за допомогою цього аналізу, щоб досягти його більшої ефективності при виявлені вроджених вад.
У лабораторії розраховують індивідуально для кожної жінки ризик народження дитини з вродженими вадами, виходячи з рівня трьох або більше компонентів крові, віку, ваги та расової належності жінки, а також з того, чи хворіє вона на діабет і приймає інсулінове лікування. Ці три останні фактори впливають на рівень МСАФП.
У який період вагітності роблять потрійний аналіз?
Цей вид аналізу найчастіше роблять між 16 і 18 тижнями після останньої менструації. Результати аналізу зазвичай бувають готові через 1 тиждень.
Якщо аналіз показує відхилення від норми, чи означає це, що дитина має вроджені вади?
Ні. За допомогою цього аналізу неможливо діагностувати вроджену ваду, він тільки вказує на підвищений ризик її виникнення. Позитивний результат означає тільки, що треба пройти додаткові обстеження. Серед 1000 жінок, які здають потрійний аналіз крові, 100, як правило, отримують результат з відхиленням від норми. І тільки у 2-3 з цих жінок народжуються діти з вродженими вадами.
Для більшості жінок позитивний результат аналізу означає тільки те, що плід або більший, або менший за віком, ніж вважалось до аналізу. Оскільки, показники норми аналізу різні в різні строки вагітності, дуже важливо знати точний гестаційний вік плоду. Якщо в цьому є труднощі, можна скористатись ультразвуковою діагностикою. Ще один типовий випадок, коли результати показують відхилення від норми, – це вагітність з більш як одним плодом (двійня, трійня, і т.п.)
Жінка, яка не знає, що більшість вагітних з позитивними результатами народжують здорових немовлят, може пережити сильне хвилювання, яке є по суті безпідставним.
При яких проблемах з плодом потрійний аналіз крові показує відхилення від норми?
Дефекти невральної трубки (ДНТ). Підвищений рівень одного з вимірюваних компонентів – МСАФП – викликає припущення про підвищений ризик дефектів невральної трубки. Невральна трубка – це частина ембріону, з якої розвивається головний та спинний мозок. В результаті неправильного зрощення невральної трубки через 4 тижні після запліднення можуть виникнути такі аномалії, як spina bifida (спино-мозкова кила) та аненцефалія. З цими вадами в нашій країні щорічно народжується 2,500 дітей.
Spina bifida, яку ще часто називають “відкритий хребет”, є вродженою аномалією хребта. У більшості випадків цього дефекту спинний мозок мальформований і випинається назовні, що призводить до різних ступенів паралічу ніг та порушень функцій сечового міхура і кишківника. При аненцефалії верхній кінець невральної трубки не закривається, що призводить до тяжких мальформацій мозку та черепу. Немовлята з аненцефалією не виживають.
Причини виникнення ДНТ не вивчені до кінця. Вчені вважають, що ці мальформації викликані генетичними факторами і несприятливим впливом навколишнього середовища. Від 90 до 95% дітей з ДНТ народжується в батьків, які не мають сімейного анамнезу цих захворювань.
Дослідження показують, що ризик ДНТ можна зменшити, якщо жінки репродуктивного віку вживатимуть перед і в перші тижні вагітності фолієву кислоту (вітамін В). Відповідно до рекомендацій Інституту Медицини та організації March of Dimes усім жінкам дітородного віку необхідно щоденно вживати 400 мікрограмів фолієвої кислоти перед заплідненням та у перші місяці вагітності. Жінки повинні притримуватись здорового харчування, включаючи продукти, багаті на фолієву кислоту (апельсиновий сік, боби, листкові зелені овочі, збагачені злакові сніданки) і щоденно приймати полівітаміни. Це єдиний правильний для жінки спосіб отримувати таку кількість фолієвої кислоти, яка їй необхідна.
Підвищення рівня МСАФП може відбутись також внаслідок деяких рідкісних дефектів черевної стінки, нирок або кишківника плоду. У рідких випадках рівень МСАФП високий в результаті вмирання або смерті плоду.
Синдром Дауна. Низькі показники МСФАП та естріолу, і при цьому високий рівень ХЛГ, вказують на підвищений ризик синдрому Дауна. Сьогодні один з вісімсот немовлят народжується з синдромом Дауна. Хворі діти мають характерні риси обличчя, розумове відставання, серцеві аномалії та інші відхилення.
Ризик синдрому Дауна та інших хромосомних захворювань підвищується пропорційно вікові жінки. Зазвичай, вагітним жінкам віком 35 і більше років, пропонують пренатальне обстеження – амніоцентез або біопсію хоріоїдних волокон, щоб діагностувати, а частіше, виключити ці хвороби. Якщо при потрійному аналізі крові виявляється, що жінка до 35 років, має такий самий ризик, як жінки в 35 і старші (приблизно одна на 270), їй також рекомендують пройти подальше пренатальне обстеження.
Яке подальше обстеження рекомендують, якщо потрійний аналіз крові показує відхилення від норми?
При позитивних результатах потрійного аналізу наступним кроком, як правило, є ультразвукова діагностика (УЗД). При УЗ обстеженні використовують звукові хвилі для отримання зображення плоду. Ультразвукова діагностика допоможе встановити гестаційний вік плоду або покаже, що жінка виношує двійню. Якщо відхилення від норми при трійному аналізі виникло в результаті однієї з цих причин, ніякі подальші обстеження не потрібні. При ультразвуковому обстеженні можуть також бути виявлені деякі серйозні вроджені вади.
Якщо УЗД не дала пояснення позитивним результатам аналізу, рекомендується пройти додаткові тести. При підвищеному ризику синдрому Дауна, в результаті потрійного аналізу крові, лікар порадить жінці зробити амніоцентез. Під час цього обстеження лікар через черевну стінку вводить в матку тонку голку і відбирає кілька чайних ложок амніотичної рідини. Ця рідина містить клітини плоду. Саме їх обстежують на синдром Дауна або інше хромосомне захворювання. Амніоцентез дає 99%-у точність при діагностуванні, а частіше виключенні, синдрому Дауна.
Коли потрійний аналіз показує підвищений ризик народження дитини з ДНТ, лікар може рекомендувати жінці пройти УЗД високої точності (2-го рівня), амніоцентез або обидві процедури разом. УЗД високої точності черепу, спинного мозку та інших органів плоду допомагає точно виявити, або виключити, серйозні ДНТ і встановити ступінь тяжкості їх проявів, але таке обстеження можливе тільки у великих медичних закладах. Якщо такий вид обстеження є недоступним, або якщо потрібна більш детальна інформація після проходження УЗД, часто пропонують амніоцентез, щоб встановити рівень АФП в амніотичній рідині. Коли рівень АФП, і усі інші причини виключені, дуже ймовірно, що плід має ДНТ. Після того, як зроблено амніоцентез, клітини плоду перевіряють на хромосомні аномалії, які інколи супроводжують ДНТ або дефекти черевної стінки.
Які переваги дає потрійний аналіз крові?
Для переважної більшості жінок, обстеження дає впевненість в тому, що їхні майбутні діти не мають вроджених вад. Результати також допомагають жінці стежити за нормальним перебігом вагітності. Наприклад, встановлення точного гестаційного віку плоду допомагає перевірити, чи його розвиток відповідає нормі. Або завдяки виявленню кількох плодів, встановлюється спеціальний спосіб спостереження вагітності.
При встановленні діагнозу або припущенні ДНТ або інших захворювань, подружжя має можливість обговорити з кваліфікованими спеціалістами свої подальші дії. Вони можуть спланувати пологи у спеціально обладнаному медичному центрі, з тим щоб дитина могла, в разі потреби, отримати необхідне лікування або хірургічне втручання зразу після народження.
У 1991 році за підтримки March of Dimes Девід Б. Шуртлеф із своїми колегами в Університеті Вашінгтону (Сіетлі) провели дослідження, яке показало, що кесарів розтин, зроблений до настання пологів, облегшує прояви паралічу в дітей з spina bifida. Якщо пренатально був поставлений аналіз spina bifida, жінка може обговорити із своїм лікарем можливість кесаревого розтину.
У деяких випадках буває важко знайти причину позитивних результатів потрійного аналізу. Їх пов’язують з такими відхиленнями, як від’єднання плаценти, передчасні пологи або мала вага дитини при народженні. В результаті ще одного дослідження за підтримки March of Dimes, проведеного Ернестом Хуком з колегами в Університеті Каліфорнії в Беркeлі, було виявлено, що підвищений рівень МСАФП без встановленої причини може бути пов’язаний із підвищеною загрозою смерті плоду в пізній вагітності. Хоча у більшості жінок з підвищеним рівнем МСАФП народжуються живі діти, зроблене відкриття допоможе лікарям запобігти деяким випадкам смерті плоду, провівши ретельне обстеження таких вагітностей у третьому триместрі. Якщо є ознаки ускладнень, в деяких випадках життя дитини може бути врятоване завдяки раннім пологам.
Жінки, старші 35 років, можуть скористатись потрійним аналізом крові у зв’язку з ймовірністю синдрому Дауна перед тим, як вирішать робити чи ні амніоцентез. Так як, при амніоцентезі виникає незначний ризик спонтанного переривання вагітності, деякі жінки намагаються, при можливості, уникнути цієї процедури. Якщо результати потрійного аналізу покажуть, що жінка має низький ризик народження дитини із синдромом Дауна, вона може відмовитись від амніоцентезу. Однак при цьому важливо не забувати, що потрійний аналіз, на відміну від амніоцентезу та біопсії хоріоїдних волокон, не дає можливості виявляти інші хромосомні порушення, так само як і діагностувати чи виключати синдром Дауна.
Переглянуто редакційною колегією I.B.I.S.: 17/10/2000
Дивіться також:
Ультразвукова діагностика
(Ultrasound Test in Pregnancy)
Т. Сороцька
Спеціаліст з інформаційного забезпечення
Українсько-Американської Програми запобігання вродженим вадам розвитку
Ультразвукове діагностування (УЗД) – це техніка, при якій використовують звукові хвилі для отримання зображення дитини (плоду) в утробі матері. УЗД менш шкідливе, ніж рентген, тому що базується на звукових хвилях, а не на променевому випромінюванні. УЗД стало надзвичайно важливою складовою пренатального догляду. Завдяки йому лікарі отримують інформацію, необхідну для планування спостереження за вагітною і поліпшення результату вагітності. Сьогодні у США ультразвукове діагностування проходять 70% вагітних жінок.
Як діє УЗД?
Ультразвуковий пристрій діє за допомогою звукових хвиль, які відштовхуються від тіла плоду. Відлуння, яке при цьому виникає, перетворюється у зображення на моніторі – сонограму. Цю техніку інколи називають сонографією.
Як проводять УЗД?
При скануванні вагітності звукові хвилі спрямовують за допомогою ручного пристрою (трансдьюсеру), проводячи ним назад і вперед по животі вагітної. Щоб отримати чіткіше зображення, на живіт наносять гель; можуть, також, попросити жінку не спорожнювати сечовий міхур перед обстеженням. Звичайне (рутинне) обстеження, воно ще називається базовим або обстеженням І-го рівня, займає від 15 до 20 хвилин. При ньому перевіряють термін вагітності, ріст і розмір плоду, кількість плодів, розташування плаценти, і дивляться, чи немає великих вроджених вад. Якщо виникає підозра вроджених вад, пацієнтку можуть направити на більш детальне обстеження (спрямоване або ІІ-го рівня), при якому використовують складніше ультразвукове обладнання, і яке може тривати від 30 хвилин до кількох годин. Деякі великі медичні центри зараз мають нове ультразвукове устаткування, яке дає тримірний, майже такий чіткий, як фотографія, вигляд плоду (3-D УЗД). Таке обладнання теж можуть використовувати, коли є підозра вроджених вад. УЗД – безболісна процедура, хоча деякі жінки скаржаться на незручність через переповнений сечовий міхур. У ранній період вагітності матка та фаллопієві труби розміщені ближче до вагіни, ніж до поверхні живота. Якщо виникає потреба в діагностиці у малий строк вагітності, звукові хвилі можуть спрямовувати за допомогою зонда, вставленого у вагіну; така техніка називається трансвагінальним УЗД.
Коли використовують УЗД?
УЗД має широке використання при обстеженні вагітності. Воно дає відповідь на безліч запитань, найважливіші з яких:
- Виключення або підтвердження підозрюваної ектопічної вагітності. УЗД може використовуватись для діагностики позаматкової вагітності, розміщеної у фаллопієвих трубах або в черевній порожнині.
- Підтвердження наявності більш, як однієї дитини. УЗД покаже, чи вагітна жінка двійнею, трійнею або має більше число плодів.
- Уточнення терміну вагітності. Розмір плоду, виміряний за допомогою ультразвуку, допомагає лікарям встановити точний строк вагітності.
- Оцінка показників росту плоду. Якщо матка збільшується швидше або повільніше, ніж очікується, за допомогою УЗД можна встановити, чому це відбувається – наприклад, через надлишок амніотичної рідини або слабкий розвиток плоду.
- Оцінка вірогідності спонтанного аборту. Якщо спостерігається кровотеча на початку вагітності, або існує припущення про зупинку серцебиття та рухів плоду, УЗД допоможе встановити, чи плід мертвий, і чи очікується в жінки викидень.
- Встановлення причини кровотечі в другому чи третьому триместрі вагітності. Такі кровотечі часто спричиняються проблемами з плацентою і потребують спеціальних заходів, включаючи кесарів розтин.
- Допомога у проведенні інших видів пренатального обстеження. Якщо вагітній призначені амніоцентез або біопсія хоріоїдних ворсин, лікарі за допомогою УЗД орієнтуються щодо місця видалення клітин для цих видів аналізу.
- Діагностування деяких вроджених вад. Ультразвукове зображення може бути використане для діагностування деяких вроджених вад будови тіла, таких зокрема, як недостатня кількість ребер або spina bifida. За допомогою УЗД діагностують також деякі мальформації внутрішніх органів, наприклад, сечовивідний тракт. Спеціальний різновид УЗД, який називається ехокардіографія, дає змогу простежити рух крові через серцеві камери, шлуночки і великі кровоносні судини плоду і виявити багато видів мальформацій серця та небезпечних для життя аномалій серцевого ритму.
- Перевірка самопочуття плоду на пізній стадії вагітності. Тест, який називається “біофізичний малюнок плоду”, поряд з іншими безстресовими процедурами, включає кілька ультразвукових обстежень (спеціальний моніторинг серцебиття плоду, який призначають, коли у матері є проблеми із здоров’ям, наприклад, цукровий діабет або високий кров’яний тиск, або коли вона переношує). Ультразвукове обстеження включає спостереження за рухами плоду, в тому числі за дихальними рухами та м’язовими тонами, і вимірювання кількості амніотичної рідини.
- Вибір виду пологів. УЗД може мати важливе значення, коли треба визначити, чи є необхідність у кесаревім розтині (або С-розтині) при деяких вагітностях, зокрема, коли плід надто великий або неправильно розташований, або коли плацента блокує вихід з матки.
Чи не шкідливе УЗД?
УЗД вважається нешкідливим для матері і дитини. Ним користуються вже протягом 30 років, і ніяких загроз не було виявлено. Деякі лікарі пропонують жінкам з групи низького ризику у плановому (рутинному) порядку проходити УЗД, щоб перевірити ріст та самопочуття плоду. Хоча, залишається остаточно не вирішеним, чи цим жінкам доцільно взагалі проходити таке обстеження. У 1993 були опубліковані результати великого за обсягом дослідження, яке показало відсутність суттєвої різниці між двома групами жінок з групи низького ризику (одні мали два рутинних УЗ обстеження, інші – пройшли УЗД через якусь встановлену лікарем причину) за такими показниками, як передчасні пологи, вага дитини при народженні, серйозні ускладнення в постнатальний період, смерть новонародженого. Спираючись на ці та подібні дослідження, Американська Колегія Акушерів та Гінекологів рекомендувала проводити УЗД тільки за конкретними медичними показами, такими, які вказані вище.
Чи має протипоказання звичайне (рутинне) УЗД?
Для жінок з групи низького ризику УЗ обстеження корисне для виключення вірогідності проблем, але не дуже ефективне для їх виявлення. У даний час при рутинному УЗД знаходять тільки половину структурних вроджених вад (хоча, згідно досліджень європейських вчених, рутинне УЗД виявляє близько трьох четвертих усіх основних вад розвитку). Деякі дослідження показують, що УЗД найбільш точне, коли його проводить досвідчений спеціаліст, який працює у великому медичному закладі. При рутинному УЗД бувають випадки, коли пропускають вроджену ваду, і, навпаки, коли “знаходять” ваду, якої насправді немає. Приблизно одну з 1000 жінок групи низького ризику помилково попередили про можливість народження дитини з вадою. У таких випадках батьки встигають пережити сильну тривогу, поки подальші обстеження не покажуть, що дитина здорова.
Чи можна вилікувати захворювання, виявлені при УЗД?
Інформацію, отриману при УЗД, часто використовують, щоб внести зміни в заходи пренатального догляду і покращити шанси жінки на народження здорової дитини. Наприклад, виявлене при УЗД порушення серцевого ритму, яке загрожує життю плода, можна лікувати медикаментами ще в утробі матері. Деякі форми непрохідності сечовивідного тракту та інші мальформації лікують хірургічним способом до народження. Виявлення деяких вроджених вад або аномалій плаценти вказують на доцільність кесаревого розтину для більшої безпеки матері і дитини.
Чи є інші способи зменшити ризик вроджених вад розвитку?
УЗД та інші методи пренатальної діагностики допомагають жінці дізнатись, чи є в її дитини вроджені вади або ризик якихось інших відхилень. Знати про ці проблеми до народження дитини – означає, мати час, щоб спланувати її лікування. Існує кілька основних речей, які може зробити кожна жінка, щоб збільшити свій шанс на здорову вагітність і здорову дитину.
- Пройти медичний огляд перед заплідненням.
- Вживати щодня 400 мікрограмів вітаміну В фолієвої кислоти, починаючи це робити до настання вагітності, щоб уникнути важких вроджених вад спинного та головного мозку дитини. Найбільш надійний спосіб вживання фолієвої кислоти – це щоденний прийом мультивітамінів в поєднанні із здоровою дієтою, яка включає їжу, багату на фолієву кислоту, як наприклад, апельсиновий сік та цитрусові, зелені листові овочі, цільні злаки та збагачені сухі сніданки.
- Регулярно, починаючи від раннього строку вагітності, проходити пренатальний медичний огляд.
- Мати нормальну вагу тіла перед заплідненням і набирати від 11 до 15 кг протягом вагітності. (Показник збільшення ваги є індивідуальним для кожної жінки, тому його необхідно узгодити із своїм лікарем).
- Утримуватись від алкогольних напоїв впродовж вагітності.
- Не палити під час вагітності.
- Не вживати ніяких пігулок, навіть загальнопоширених медикаментів, без рекомендації лікаря, який знає, що ви вагітна.
Переглянуто редакційною колегією I.B.I.S.: 5/02/2002
Синдром Ангельмана
(“Радісної маріонетки” синдром)
(Angelman Syndrome)
Наталія Ігорівна Юськів
Лікар-генетик
Волинського обласного дитячого територіального медичного об’єднання
Основні діагностичні критерії:
Розумова відсталість, загальний мовний недорозвиток важкого ступеню, характерна атактична хода (“хода механічної ляльки”: крокування із зігнутими в ліктях руками), мікроцефалія, сплощена потилиця, нижній (мандибулярний) прогнатизм, протрузія язика, пароксизми немотивованого сміху, дифузні зміни на ЕЕГ.
Клінічна характеристика:
Мікроцефалія часто поєднується з гіпотрофією, моторною гіперактивністю та інверсією сну. В ранньому дитинстві відмічається затримка психомовного розвитку важкого ступеню, атаксія та в’ялі посмикування рук. У пацієнтів з синдромом Ангельмана характерний радісний вираз обличчя та приступи (рефлекторні) неадекватного сміху. Постійно відмічається глибока розумова відсталість, хворі здатні виконувати тільки прості інструкції та команди.
В ранньому дитинстві у більшості хворих з’являються епілептичні приступи. На комп’ютерній томограмі виявляють помірну атрофію кори головного мозку та розширення вентрикулярної системи.
ЕЕГ патологічно змінена. Фіксуються генералізовані піки високої амплітуди та повільні хвилі (2-3 ц. за сек.).
Ускладнення:
Пов’язані з епіприступами та атаксією; набуті ортопедичні деформації.
Етіологія:
Синдром Ангельмана відноситься до хвороб, обумовлених геномним імпринтингом (від англ. “imprint” – “відбиток”). Причиною може бути успадкована від матері делеція 15-ї хромосоми (q11-q13), батьківська дисомія 15 (в 3%-5% випадків), точкова мутація активного алеля або мутація імпринтингового центру.
Патогенез:
Основні клінічні прояви є наслідком ураження центральної нервової системи. Мікробрахіцефалія, нижня прогнатія є результатом недорозвитку мозку. Атаксія, пароксизми сміху, алексія, епіприступи та патологічна ЕЕГ є наслідком функціональної дезорганізації ЦНС.
Ризик для сибсів пробанда: приблизно 50%.
Картування генів:
ANCR (Angelman Syndrome Chromosome Region) картований на 15(q11-q13).
Профілактика:
Рекомендоване медико-генетичне консультування.
Лікування:
Раннє навчання “мові жестів” та тренування невербальних навиків може дещо покращити перспективу таких дітей в плані соціальної адаптації. Прогноз щодо розвитку мови – несприятливий. Більшість хворих не мають навиків самообслуговування і потребують сторонньої допомоги протягом цілого життя.
Номер з каталогу МІМ:
105830 Angelman Syndrome, AS
Список літератури:
- Greenberg F. Contiguous gene syndrome. Growth: Genetics and Hormones 1993;9:5-10.
- Lalande M. Parental imprinting and human disease. Annual Review of Genetics 1997;30:173-195.
- Malcom S. Microdeletion and microduplication syndromes. Prenatal Diagnosis 1996;16:1213.
- McKusick VA. Happy Puppet Syndrome. In: Mendelian Inheritance in Man: Catalogs of Autosomal Dominant, Autosomal Recessive, and X-Linked Phenotypes. 10th ed. V. 2. Baltimore: The Johns Hopkins University Press, 1992:1431.
- Williams CA, Frias JL. Angelman Syndrome. In: Buyse ML, ed. Birth Defects Encyclopedia. Dover: Center for Birth Defects Information Services, Inc., 1990:140-141.
Переглянуто редакційною колегією I.B.I.S.: 15/02/2002
Амніоцентез
(Amniocentesis)
Т. Сороцька
Спеціаліст з інформаційного забезпечення
Українсько-Американської Програми
запобігання вродженим вадам розвитку
Амніоцентез – це проста медична процедура, за допомогою якої отримують для аналізу невелику кількість амніотичної рідини, яка оточує плід. Вперше її провели в 1882 році, і з того часу виконують в третьому триместрі вагітності, щоб виміряти рівень анемії при гемолітичній хворобі плоду або дізнатись, чи легені плоду достатньо розвинуті для початку пологів. У даний час амніоцентез часто використовують в другому триместрі вагітності для діагностики, а скоріше для виключення певних вроджених вад.
У яких випадках рекомендують амніоцентез?
Амніоцентез не пропонують усім жінкам, як рутинну (звичайну, без спеціального призначення лікаря) процедуру, тому що вона пов’язана із незначним ризиком спонтанного аборту. Амніоцентез рекомендують, коли існує підвищений ризик хромосомних або генетичних вроджених вад.
За допомогою іншого пренатального тесту, який називається біопсією хоріоїдних волокон (БХВ), можна продіагностувати більше (але не усі) видів вроджених вад, ніж за допомогою амніоцентезу. БХВ роблять на ранній стадії вагітності (зазвичай, між 10 і 12 тижнями), але, при цьому, як було встановлено, виникає трохи більший ризик спонтанного переривання вагітності та інших ускладнень. Деякі дослідження вказують на те, що БХВ пов’язана, також, з дуже незначним ризиком вроджених вад, зокрема, дефектів пальців рук і ніг.
Амніоцентез рекомендують у випадках, пов’язаних із:
- Віком матері. Ризик народження дитини з певними хромосомними вродженими вадами зростає пропорційно вікові матері. Жінкам, віком 35 і старшим, більшість лікарів рекомендують пройти на вибір один з тестів на хромосомні порушення. Найбільш поширеним з таких порушень є синдром Дауна – поєднання фізичних та розумових аномалій, спричинене зайвою хромосомою. Цей синдром трапляється в 1 з 1250 дітей, народжених жінками віком від 20 до 30 років; у віці 35 років вірогідність його виникнення збільшується до 1:385, а в сорок років – до 1:106.
- Наявністю вроджених вад розвитку у попередньої дитини (попередній вагітності). Якщо у подружжя вже народжувалась дитина, або була вагітність, з діагнозом хромосомної аномалії, набору генетичних вроджених вад або дефекту невральної трубки ( див. нижче), при наступній вагітності їм можуть запропонувати пренатальне тестування.
- Іншими аномаліями в родинній історії. Подружжям, в яких не народжувалось хворих дітей, теж можуть запропонувати пройти пренатальне тестування, якщо медичні історії їхніх сімей вказують на те, що нащадки мають підвищений ризик успадкування генетичної хвороби. Пренатальне обстеження буде призначено тільки в тому випадку, якщо підозрювану хворобу можна діагностувати пренатально. Практично усі хромосомні, але не усі генетичні, порушення можна діагностувати пренатально.
- Ймовірність виникнення дефектів невральної трубки. Амніоцентез допомогає в діагностиці дефектів невральної трубки (дефекти спинного та головного мозку, включаючи spina bifida та аненцефалію). Це можливо шляхом виміру рівня речовини, яку виробляє плід, – альфа – фетопротеїну (АФП) в амніотичній рідині. Амніоцентез для вимірювання рівню АФП призначають, коли існує сімейна історія дефектів невральної трубки, або якщо результат аналізу крові матері на АФП показав, що вагітність пов’язана з підвищеним ризиком.
Як роблять амніоцентез?
В амніотичній рідині плавають живі клітини плоду. Їх видаляють із проби амніотичної рідини вагітної жінки, вирощують в лабораторії протягом одного або двох тижнів, і потім перевіряють на хромосомні та генетичні вроджені вади. Результати аналізу можна отримати, зазвичай, через тиждень або два.
В амніотичній рідині міститься також протеїн. Оскільки його можна виміряти безпосередньо, не чекаючи, коли клітини виростуть, аналіз займає усього кілька днів.
Як проходить процедура амніоцентезу?
Ульразвукове зображення дає точну картину розміщення плоду та плаценти, і це дозволяє лікарю вибрати безпечне місце для введення пункційної голки. Далі поверхню живота дезинфікують, лікар вводить в матку тонку голку і відбирає кілька чайних ложок амніотичної рідини. Після того, як проба узята, лікар за допомогою ультразвуку перевіряє серцебиття плоду.
Деякі жінки кажуть, що амніоцентез зовсім безболісна процедура, інші відчувають спазм, коли голка входить у матку, або тиснучий біль протягом кількох хвилин після забору рідини. Після амніоцентезу від одного до двох відсотків жінок скаржаться на біль, мають виділення в місці проколу або витікання рідини. Лікарі рекомендують протягом кількох годин після цієї процедури відпочивати, уникати фізичного навантаження, не піднімати важкого і не стояти протягом довгого часу.
Коли роблять амніоцентез?
Зазвичай, амніоцентез роблять між 15 і 18 тижнями після останньої менструації. У деяких медичних установах рекомендують амніоцентез на ранній стадії, а саме, між 11 та 14 тижнем після останньої менструації. Однак, вважається що ранній амніоцентез можна робити тільки експериментально, а останні дослідження показали, що він пов’язаний з більшим ризиком, ніж амніоцентез в другому чи третьому триместрі вагітності (див. нижче).
У третьому триместрі амніоцентез виконує ще кілька функцій. Його використовують не тільки для того, щоб встановити, чи легені плоду достатньо сформовані у разі необхідності передчасних пологів. Амніоцентез допомагає в діагностиці внутрішньоутробних інфекцій і може, також, бути рекомендований при передчасному розриві навколоплідної оболонки. За допомогою амніоцентезу можна також встановити ступінь анемії плоду при гемолітичній хворобі, щоб вирішити, чи необхідно для збереження життя дитини робити переливання крові.
Чи небезпечний амніоцентез?
Мільйони жінок робили амніоцентез з метою пренатальної діагностики. У 1976 році після ретельних досліджень Національний Інститут Здоров’я повідомив, що триместровий амніоцентез для пренатальної діагностики (при орієнтуванні за допомогою ультразвуку) можна вважати нешкідливим. Однак, з амніоцентезом усе таки пов’язаний незначний ризик спонтанного переривання вагітності. Згідно даних Центру Контролю та Запобігання Захворюванням, показник спонтанних абортів після амніоцентезу коливається від 1:400 до 1:200. При цій процедурі припускається також надзвичайно низький ризик внутрішньоутробної інфекції ( менше, ніж 1 випадок на 1000), яка може привести до спонтанного переривання вагітності.
В процесі досліджень встановлено, що ризик самоаборту після амніоцентезу, зробленого в першому триместрі, може бути в три рази вищий, ніж ризик при друготриместровому амніоценезі. Нещодавно канадськими дослідниками було підраховано, що ризик спонтанного аборту становить 2,6 % після раннього амніоцентезу в порівнянні з 0,8% – при амніоцентезі у другому триместрі. Вони також виявили вражаюче зростання ризику деформації ніг, яка називається клишоногістю, у новонароджених, матерям яких робили амніоцентез на ранній стадії вагітності. Ризик клишоногості зростає в десять разів після раннього амніоцентезу (1,3% проти 0,1% (1:1000) після амніоцентезі в другому триместрі). Описані випадки клишоногості після друготриместрового амніоценезу нічим не відрізняються від випадків в немовлят в США. Спираючись на це та інші дослідження, лікарі змінили своє ставлення до раннього амніоценезу. Багато з них вважають, що, якщо в першому триместрі виникає необхідність в пренатальному тестуванні, то біопсія хоріоїдних волокон є для цього більш безпечною, ніж ранній амніоцентез.
Ризик втрати вагітності в результаті амніоценезу зменшується, якщо процедуру проводить лікар з великим досвідом. Досвідчених спеціалістів, зазвичай, можна знайти у великих медичних установах. Допомогти в цьому може дільничий/ сімейний лікар або лікар-генетик.
Чи означають нормальні результати амніоцентезу, що дитина народиться здоровою?
Більш як 95% жінок з групи підвищеного ризику, які пройшли пренатальну діагностику, отримують підбадьорливу новину про те, що в їхніх дітей не виявлено хвороб, з приводу яких робилось тестування. Однак, жодний пренатальний тест не може гарантувати народження здорової дитини, так як тільки деякі з вроджених вад можуть бути виявлені пренатально. Від 3 до 4 на 100 немовлят мають вроджені вади. Точність даних амніоцентезу при діагностуванні хромосомних аномалій становить від 99,4 до 100 відсотків.
Чи виліковні вроджені вади, діагностовані за допомогою амніоцентезу?
На даний час лікарі здатні продіагностувати набагато більше вроджених вад, ніж вони можуть вилікувати пренатально. Однак, завдяки досягненням пренатальної терапії стало можливим лікування деяких вроджених порушень ще до народження. Прикладом може бути випадок, коли дітей з біотиновою залежністю та метилмалонічною ацидемією (спадкові небезпечні для життя порушення хімічних процесів в організмі), виявленими за допомогою амніоцентезу, лікували в утробі матері, і вони народились здоровими.
Навіть якщо плід має ваду, для якої поки що не існує пренатального лікування, пренатальна діагностика допоможе батькам підготуватись морально і обговорити із своїм лікарем оптимальний час, місце та спосіб пологів з тим, щоб зразу після народження дитини усі необхідні спеціалісти були в їхньому розпорядженні.
Кому потрібно робити амніоцентез?
Батьки майбутньої дитини самостійно вирішують, проходити чи ні пренатальну діагностику, після обговорення цього питання із спеціалістами. Цінну допомогу батькам у прийнятті правильного рішення щодо пренатального обстеження та в інших питаннях, пов”язаних з народженням дитини, можуть надати генетики-консультанти, терапевти, консультанти з релігійних та етичних проблем.
Деякі тести, як напр., БХВ, проводять у ранній період вагітності, і необхідність вирішити, проходити обстеження далі, чи відмовитись від нього, вимагає від лікаря ретельно продуманого і підготовленого обговорення цього питання з пацієнтами. Це ще один доказ на користь того, що кожна жінка повинна на ранній стадії вагітності стати на медичний облік, і відвідати лікаря ще до настання вагітності.
Чи є способи зменшити ризик вроджених вад розвитку?
Існують кілька основних речей, які може зробити кожна жінка, щоб збільшити свій шанс на здорову вагітність і здорову дитину.
- Спланувати вагітність, відвідавши лікаря ще до заплідненням.
- Вживати щодня 400 мікрограмів фолієвої кислоти (кількість, яка зазвичай міститься в полівітамінах), почавши це робити до настання вагітності і продовжуючи протягом першого триместру, щоб запобігти дефектам невральної трубки.
- Регулярно, починаючи від раннього строку вагітності проходити пренатальний медичний огляд.
- Харчуватись різноманітно і поживно.
- Мати нормальну вагу тіла ( бути ні надто повною, ні надто худою) перед заплідненням.
- Утримуватись від алкогольних напоїв впродовж вагітності.
- Не палити під час вагітності.
- Не вживати ніяких пігулок, навіть загальнопоширених медикаментів, без рекомендації лікаря, який знає, що ви вагітна.
Переглянуто редакційною колегією I.B.I.S.: 5/02/2002
Дивіться також:
Синдром амніотичних тяжів
(Amniotic Constricting Band Syndrome)
Лікар ультразвукової діагностики
Рівненського обласного клінічного лікувально-діагностичного центру
Включення:
Аномальні складки шкіри, Адам-комплекс, амніохоріонічні мезобластичні фіброзні тяжі, амніогенні складки, ампутаційний комплекс при амніотичних тяжах, вроджені кільцеві тяжі, вроджений констриктивний синдром, вроджені кругові тяжі і вроджені поперечні дефекти.
Частота:
Синдром зустрічається з частотою 1:1200 – 1:15000 новонароджених.
Співвідношення статей: Ч1 : Ж1
Етіопатогенез:
Синдром амніотичних тяжів – спорадичний стан. Незважаючи на те, що існує декілька теорій, які пояснюють генез синдрому, найбільш розповсюдженою є думка про ранній розрив амніону з утворенням мезодермальних тяжів, що виходять з хоріонічного боку амніону, та їх проникнення в тіло плоду, що, в свою чергу, призводить до ампутації, звуження і вторинних постуральних деформацій внаслідок іммобілізації плоду. Було відмічено, що чим раніше виникають дефекти амніону, тим більше виражені ушкодження плоду. Розрив амніону на першому тижні вагітності призводить до черепно-лицевих деформацій, дефектів внутрішніх органів, тоді як при його розриві в ІІ триместрі вагітності у плода утворюються перетяжки з ампутацією кінцівок і пальців. Альтернативою є думка про те, що синдром амніотичних тяжів – це результат пошкодження, що призводить до типових вад розвитку, пов’язаних з ектодермальними і мезодермальними порушеннями. Судинний фактор також може відігравати роль в патогенезі зовнішнього дефекту. Розвиток синдрому, за даними літератури, можливий після амніоцентезу.
Клінічна картина:
Найчастіше при синдромі амніотичних тяжів зустрічаються такі аномалії, як множинні асиметричні дефекти кінцівок, черепно-лицеві дефекти, вісцеральні дефекти (таб. 1).
Таб. 1. Аномалії, поєднані з синдромом амніотичних тяжів
– Кругові перетяжки кінцівок і пальців; – Ампутація кінцівок і пальців; – Псевдосиндактилія; – Аномальні форми шкірних виростків; – Мавп’ячі складки; – Клишоногість двобічна. |
дефекти: – Енцефалоцеле множинне, асиметричне; – Аненцефалія; – Лицеві розщілини: губи і піднебіння; – Виражені деформації носа; – Асиметрична мікрофтальмія; – Відсутність або недосконалий тип кальцифікації черепа. |
– Гастрошизис; – Омфалоцеле. |
В 77% випадках виявляються множинні аномалії. При енцефалоцеле процес часто буває асиметричний і може бути множинним. Подібна картина майже патогномонічна для синдрому амніотичних тяжів, так як зазвичай ця аномалія буває ізольованою і локалізується по середній лінії. Синдром амніотичних тяжів може бути представлений вираженими аномаліями обличчя, у тому числі анофтальмією, мікрофтальмією і хаотичними множинними розщілинами. Кругові деформації включають два види пошкоджень: ті, що можуть бути пов’язані з руйнівним ефектом тяжів, наприклад, ампутації і кругові перетяжки, і пов’язані з іммобілізацією плоду, наприклад, клишоногість, яка зустрічається у 2/3 всіх випадків. Внутрішні органи не змінені, однак, якщо амніон розривається під час фізіологічної евісцерації вмісту черевної порожнини, мезодермальні тяжі можуть бути причиною таких дефектів розвитку передньої черевної стінки, як гастрошизис і рідше омфалоцеле. У деяких випадках виявляється кифосколіоз.
Фіброзні тяжі, що відходять від поверхні плаценти, часто помітні як проникаючі у тіло плода на рівні виникаючих дефектів. У деяких плодів в результаті амніотичного розриву утворюється зв’язок між порожниною амніону (екстраамніотична вагітність) і екстраембріональним целолом. Плід може вийти з порожнини амніону. Ріст амніону припиняється, і він виявляється після народження дитини у вигляді невеликого мішка, зв’язаного з пуповиною.
На схемі 1 представлено клінічні підходи до оцінки внутрішньоматкових тяжеподібних структур.
Схема 1. Клінічні підходи до оцінки внутрішньоматкових тяжеподібних структур
 Пренатальна діагностика:
Пренатальна діагностика:
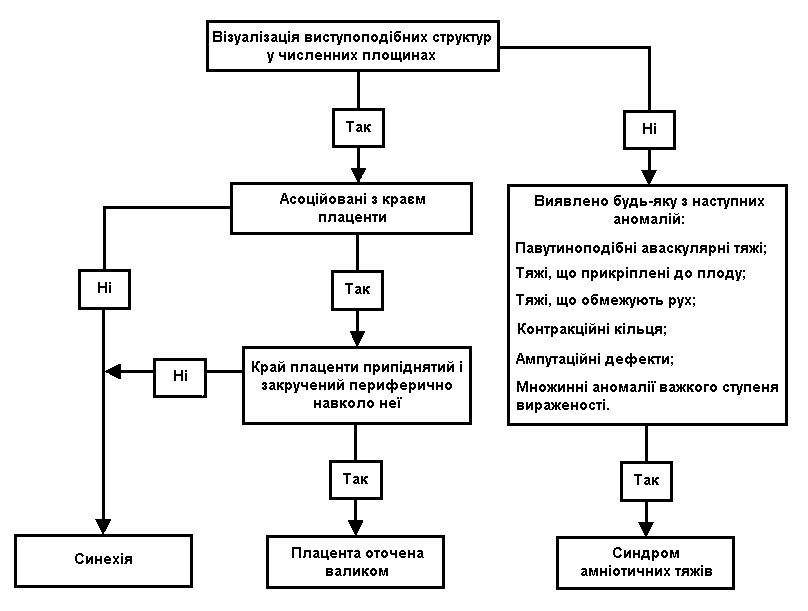
Синдром являє собою широкий спектр аномалій, що проявляються у кожному випадку по-різному. Підозра може виникнути, коли нараховується дві аномалії або більше, вказані у таблиці 1. Асиметричне або множинне енцефалоцеле у поєднанні з ампутаціями, дефектами передньої черевної стінки і постуральними деформаціями підвищує вірогідність цієї патології.
Деколи амніотичний тяж можна побачити як лінійну структуру, вільно плаваючу в навколоплідних водах і зв’язану з тілом плода. Однак маловіддя може ускладнити виявлення тяжів. Труднощі виникають при диференційному діагнозі двох станів, які можуть бути причиною візуалізації лінійних ехоструктур, що пересікають порожнину амніону: хоріоамніонічний розподіл і внутрішньоматкові синехії. У цих випадках необхідне сканування плода, спрямоване на пошук з’єднання перегородок з його тілом. Синехії його не мають і не викликають структурних аномалій.
Прогноз:
Прогноз залежить від вираженості аномалій. Їх спектр варіює від незміненого інтелекту у дітей, у яких відмічаються незначні констриктивні дефекти пальців, до множинних важких аномалій, несумісних з життям.
Акушерська тактика:
Необхідне каріотипування плода, оскільки множинні вроджені аномалії можуть бути результатом хромосомних порушень. При виявленні патології до початку періоду життєздатності плода оптимальним методом є переривання вагітності. При встановленні діагнозу після початку періоду життєздатності плода акушерська тактика не змінюється до тих пір, поки не виявляться аномалії, несумісні з життям (аненцефалія, наприклад). У цих випадках оптимальним є обговорення питання з родичами про переривання вагітності на будь-якому її терміні.
Номер з каталогу МІМ:
217100 Constricting Bands, Congenital
Література:
- Козлова С.И., Демикова Н.С. и др. Наследственные синдромы и медико – генетическое консультирование. Л.: Медицина,1987.- C. 28-29.
- Ромеро Р., Пилу Дж. и др. Пренатальная диагностика врожденных пороков развития плода /Пер. с англ.- М.: Медицина,1974.- C.412-414.
- Jones KL. Smith’s recognizable patterns of human malformations. W.B.Sauders Company, California. 1981:636-639.
Переглянуто редакційною колегією I.B.I.S.: 11/03/2002
Спінальна аміотрофія Вердніга-Гоффмана
(Werdnig-Hoffmann Spinal Muscular Atrophy)
Лікар-невролог
Рівненської обласної клінічної дитячої лікарні
Включення:
Хвороба “кволої дитини”.
Визначення:
Основою захворювання є дегенерація рухових клітин передніх рогів спинного мозку.
Основні діагностичні критерії:
- Аутосомно-рецесивний тип успадкування.
- Дебют захворювання – пренатальний період та перші 6 місяців життя.
- Симптомокомплекс “кволої дитини” – генералізована м’язова гіпотонія, слабкість м’язів тулуба та переважно проксимальних відділів кінцівок, фасцикуляції.
- Затримка моторного розвитку.
- Наявність в біоптатах м’язів групувань дрібних круглих волокон, гіпертрофованих волокон І типу та атрофованих волокон І та ІІ типів.
- Ознаки денервації при ЕМГ-дослідженні.
- Швидке прогресування хвороби.
Клінічна картина (перебіг хвороби):
Під час вагітності мати відмічає кволі порухи плоду. Відразу після народження – м’язова гіпотонія, слабкість, зниження або відсутність сухожилкових рефлексів. В окремих випадках ознаки бульбарних розладів: слабкий крик, мляве смоктання, дисфагія, фібриляції м’язів язика. Характерне положення у вигляді розспластаної жаби – з відведеними стегнами та зігнутими колінами. Ураження міжреберних м’язів та діафрагми призводить до виникнення парадоксального дихання. На протязі перших місяців життя відмічаються часті аспірації, респіраторні інфекції. У віці 6 місяців у всіх хворих спостерігається затримка моторного розвитку. Об’єм активних рухів різко обмежений. Інтелект збережений.
Дані лабораторно-інструментальних досліджень:
- Електроміографія – спонтанна біоелектрична активність і синхронізації біопотенціалів (“ритм частоколу”), а у важких випадках – “біоелектричне мовчання”.
- Біопсія м’язів – типова “пучкова” атрофія м’язових волокон.
- Патоморфологічне дослідження – дифузні дегенеративні зміни в передніх рогах спинного мозку, рухових ядрах черепних нервів (V, VI, VII, IX, X, XI, XII пари), хроматолізис, кулеподібне набухання та/ або зморщування моторних клітин, дифузна мікрогліальна та/ або астроцитарна проліферація, часто з утворенням щільних гліальних волокон. Світлова мікроскопія – виявлення атрофованих волокон І та ІІ типів. Характерно скупчення маленьких круглих волокон, які чергуються з гіпертрофованими волокнами, переважно І типу. Електронна мікроскопія: дифузне ушкодження міофібрил, фрагментація саркоплазматичної сітки, зміни Z-смуг, скупчення ядер міоцитів.
- Біохімічний аналіз крові – постійна креатинурія при нормальній активності м’язових ферментів в сироватці крові.
- ДНК-діагностика.
Тип успадкування:
Аутосомно-рецесивний. Частота гетерозиготного носійства складає 1 : 6-80. Ген локалізований на 5q11.2-13.3.
Частота: 1 : 6000 новонароджених.
Співвідношення статей: Ч1 : Ж1.
Вік прояву:
Пренатальний період, період новонародженості, ранній дитячій вік.
Пренатальна діагностика:
- Відставання матки в рості.
- Пізні, кволі рухи плоду.
- Гідрамніон.
- Зміни кількості ребер плода.
- ДНК-діагностика.
Лікування:
- Психологічна підтримка хворих.
- Медикаментозна терапія не існує.
- Проводять випробовування нейротрофічних факторів (циліарного нейротрофічного фактору CNTF і мозкового нейротрофічного фактору BDNF), блокаторів глутаматних рецепторів, інгібіторів супероксиддисмутази.
- Підсадка NAIP-гена в клітини передніх рогів спинного мозку за допомогою генетично змодульованого вектора поліовіруса.
- Симптоматичне лікування м’язової слабкості.
- Механічні пристосування (комірці, бинтування зап’ястків).
- Моторизовані засоби.
- Лікувальна фізкультура (ЛФК).
Номер з каталогу МІМ:
253300 Spinal Muscular Atrophy, Type I; SMA1
Література:
- Козлова С.И., Демикова Н.С., Семанова Е., Блинникова О.Е.. Наследственные синдромы и медико-генетическое консультирование.- М.: Практика, 1996.- С. 174-178.
- Наследственные болезни нервной системы: Рук-во для врачей /Под ред. Ю.Е. Вельтицева, П.А. Темина. – М.: Медицина, 1998.- С. 290-294, 333-343.
- Самуэльс. Неврология.- М., 1997.
- Шанько Г.Г., Бондаренко Е.С. Неврология детского возраста.- Минск, 1990.
- Spinal Muscular Atrophy Type, I; SMA1. OMIM 253300.
Переглянуто редакційною колегією I.B.I.S.: 5/11/2002
Синдром Альстрема
(Alstrom Syndrome)
Д.Р. Ахмеджанова
Інформаційний спеціаліст Хмельницького ОМНІ-Центру
Що таке синдром Альстрема?
Синдром Альстрема – це рідкісне спадкове захворювання, перші ознаки та симптоми якого з’являються у ранньому віці. При цьому спостерігається ураження багатьох систем. Вперше його описав шведський офтальмолог Альстрем (С.Н. Alstrom) у 1959 році.
Інша назва синдрому Альстрема – синдром Альстрема–Халлгрена (Alstrom-Hallgren syndrome).
Як часто виникає цей синдром?
Цей синдром є доволі рідкісним: в усьому світі відомо про 300 випадків. Хоча зазвичай цей синдром зустрічається рідко, найбільшого поширення він набув серед акадійців, які проживають у Новій Шотландії (провінція Канади) та у Луїзіані (штат США).
Чому виникає синдром Альстрема?
Виникнення синдрому Альстрема спричиняє мутація гену ALMS1, який знаходиться на плечі 2p13 2-ї хромосоми.
Як успадковується синдром Альстрема?
Цей стан успадковується за аутосомно-рецесивним типом, тобто для виникнення захворювання необхідна мутація двох копій цього гена. Кожен з батьків дитини із синдромом Альстрема є носієм однієї копії мутованого гена і кожна їх дитина має 25% ризик успадкування обох генів. Носії не є хворими і не мають жодної ознаки або симптому хвороби.
Чи необхідна родині консультація лікаря-генетика?
Так. Лікар встановить діагноз, порекомендує методи допологової діагностики (у т.ч. застосування найсучасніших методів молекулярної діагностики) для запобігання повторного захворювання при наступній вагітності.
Які основні ознаки та симптоми синдрому Альстрема?
Ознаки та симтоми синдрому Альстрема різняться за ступенем важкості і не обов’язково у всіх хворих проявляються всі характерні ознаки.
Це прогресуюче генетичне захворювання характеризується ожирінням, глухотою, проблемами із зором у дитинстві, а також діабетом 2 типу та порушенням ниркової функції у дорослому віці.
Найпершими ознаками синдрому у ранньому віці часто є сильна чутливість до світла (фотофобія) та спонтанні рухи очей (ністагм). Уже у ранньому малюковому віці може розвинутись застійна серцева недостатність внаслідок кардіоміопатії, протягом першого року життя спостерігається зайва вага. Пізніше можуть також спостерігатись ураження інших систем органів, що призводить до сліпоти, порушення слуху, діабету 2 типу, серцевої недостатності, хвороб печінки, порушень сечо-статевої системи, легеневого фіброзу та ниркової недостатності.
У деяких випадках також можуть спостерігатись гіпотиреоз, порушення статевого розвитку, низький зріст, легка або помірна затримка розумового розвитку та такі ускладнення діабету 2 типу, як гіперліпідемія (високий рівень ліпідів) та атеросклероз. Серед проблем із зором можна виділити дистрофію сітківки. У деяких хворих спостерігається так звана акантокератодермія, тобто потовстішання, потемніння шкіри на складках.
Ендокринні порушення у жінок включають:
- зниження рівня гонадотропінів плазми;
- гірсутизм (надмірне оволосіння);
- порушення розвитку молочних залоз;
- полікістоз яєчників, передчасний статевий розвиток (пубертат у 8 років);
- порушення менструального циклу.
Помірні симптоми – нетримання сечі та симптоми пов’язані з періодичним інфікуванням.
У хворих є ризик розвитку панкреатиту.
Інші прояви – сколіоз та кіфоз різного ступеня вираженості зустрічаються у третини хворих і носіїв синдрому Альстрема.
Нейросенсорна глухота в перші 10 років життя у 70% хворих прогресує від помірної до тяжкої (40-70дБ). Від 10 до 30 років спостерігається значне зниження слуху.
Затримка розумового розвитку, як правило, не сягає важкого ступеня. Ці діти мають труднощі в навчанні, сприйнятті та мовному розвитку, затримку формування моторних навичок. У 30% відзначається нездатність до навчання.
Якщо є підозра на синдром Альстрема, то які обстеження необхідно зробити?
При підозрі на наявність синдрому Альстрема необхідно проведення наступного переліку досліджень:
- Ретельний контроль антропометричних даних: зріст, вага, індекс маси тіла.
- Офтальмологічне обстеження.
- Консультація кардіолога з проведенням ЕКГ, Ехо-КГ.
- Консультація сурдолога з проведенням аудіометрії (якщо пацієнт віком більше 4 років).
- Консультація ендокринолога, що зазвичай рекомендує визначення глюкози, інсуліну, холестерину.
- УЗД нирок та органів малого тазу.
- Печінкові проби (АЛТ, АСТ) та УЗД печінки та жовчних шляхів.
- Визначення рівня глюкози натще, а після 6 років проведення тесту на толерантність до глюкози.
Які методи лікування синдрому Альстрема сьогодні використовують?
Специфічного лікування не розроблено, тому терапія симптоматична:
- При фотодисфорії – використання кольорових лінз.
- При ожирінні – лікувальне харчування за загальними принципами, регулярне фізичне навантаження.
- При нейросенсорній глухоті – хірургічна корекція у пацієнтів з ексудативним отитом. Цифрові слухові апарати приносять полегшення деяким хворим.
- При цукровому діабеті – лікування за загальними принципами.
- При урологічних порушеннях – деяким хворим потрібен сечовий катетер, щоб уникнути труднощів з сечовиділенням.
- При хронічній обструктивній хворобі легень – лікування відповідно до загально прийнятих протоколів.
- При гіпотиреозі – терапія тироксином.
Переглянуто редакційною колегією I.B.I.S.: 25/06/2005
Дивіться також:
- Синдром Альстрема (інформація для спеціалістів)
Синдром Альстрема
(Alstrom Syndrome)
Лікар-генетик
Миколаївської обласної дитячої лікарні
Синдром Альстрема, що характеризується пігментною дегенерацією сітківки, ожирінням, прогресуючою нейросенсорною глухотою, дилятаційною кардіоміопатією, цукровим діабетом та нефропатією, описаний у 1959 р. шведським офтальмологом С. Alstrem.
Частота:
Описано біля 300 випадків синдрому Альстрема. Частота синдрому Альстрема вища у французько-акадійській популяції та серед деяких інших географічних та етнічних ізолятів.
Етіологія:
Єдиний ген, відомий на сьогодні, пов’язаний із синдромом Альстрема – це ALMS1. Підтверджується мутація молекулярним методом у 20-45% пацієнтів. Ген картований 2р13.
Основні діагностичні критерії:
Діагноз синдрому Альстрема значною мірою базується на наявності кардинальних клінічних ознак, які можуть проявитися протягом всього життя. Кардинальними ознаками вважаються:
- Пігментна дегенерація сітківки з ністагмом і фотодисфорією.
- Ожиріння.
- Прогресуюче ураження слуху.
- Дилятаційна кардіоміопатія (ДКМП).
- Синдром інсулінорезистентності (метаболічний синдром, синдром Х, чи метаболічний синдром Х).
Клініка:
Характерна значна варіабельність клінічної картини навіть серед сибсів.
Діти при народженні мають нормальну масу тіла, але протягом 1-го року життя з’являється ожиріння. Індекс маси тіла більше 95 центилей. Для дітей з синдромом Альстрема характерним є швидкий темп росту до періоду пубертату та випередження кісткового віку. Раннє закриття зон росту зумовлює низький кінцевий зріст (<5 перцентиля) у 85% пацієнтів. Згодом формується сколіоз чи кіфоз.
Вже з перших місяців життя спостерігаються ністагм та фотодисфорія. Застосування електроретинографії дозволяє виявити прогресуюче погіршення функції паличок та колбочок. Також діагностується пігментна дегенерація сітківки. Огляд очного дна в перші роки може не виявити патології, або може показати блідий очний диск і звуження судин сітківки. Пізніше з’являються прогресуюча дистрофія нейроепітелію з атрофією та пігментною інфільтрацією внутрішніх шарів сітківки, які до 7-річного віку можуть призвести до сліпоти, розвитку катаракти.
Нейросенсорна глухота в першому десятиріччі у 70% хворих прогресує від помірної до тяжкої (40-70 дБ). Від 10 до 30 років спостерігається значне зниження слуху.
Дилятаційна кардіоміопатія (ДКМП) з маніфестацією у дитячому чи юнацькому віці спостерігається більш ніж у 60% випадків. На деяких етапах життя призводить до виникнення серцевої недостатності (СН) та смерті. Зазвичай клініка дилятаційної КМП та, як її результат, СН залежить від віку їх прояву. Близько 80% хворих переносять дилятаційну КМП у молодому віці. З них 10% мають рецидиви, які прогресують у більш пізньому віці. Схильність до інфантильної КМП можлива тому, що деякі діти мають недіагностований синдром Альстрема. При аутопсії міокарда виявляються фіброзні зміни.
Синдром інсулінорезистентності (метаболічний синдром, синдром Х, чи метаболічний синдром Х) діагностується при наявності 3-х із 4-х наступних критеріїв:
- інсулінорезистентність (від гіперінсулінемії до непереносимості глюкози);
- дисліпідемія (висока концентрація в сироватці загального холестерину, ліпопротеїдів низької щільності (ЛПНЩ), тригліцеридів та низька концентрація ліпопротеїдів високої щільності – ЛПВЩ);
- підвищений артеріальний тиск (АТ);
- ожиріння, переважно тулуба.
Часто з інсулінорезистентністю пов’язаний чорний акантоз (acanthosis nigricans).
Затримка розумового розвитку описана у деяких пацієнтів, хоча як правило, не сягає важкого ступеня. Ці діти мають труднощі в навчанні, сприйнятті та мовному розвитку, затримку формування моторних навичок. У 30% відзначається нездатність до навчання.
Таблиця 1. Головні клінічні особливості, вік маніфестації і частота прояву синдрому Альстрема
(в середньому – 2,5 р) |
||
Інфантильна Юнацька |
3 тиж. – 4 міс. Підлітки і більше 30 р. |
35% 25% |
дорослий |
||
23% чоловіків |
||
у всіх хворих |
||
Цукровий діабет – це результат резистентності тканин до дії інсуліну. Діабет ІІ типу супроводжується змінами на шкірі – acanthosis nigricans – оксамитові плями гіперпігментації в ділянках підвищеної пітливості.
Гіпогонадотропний гіпогонадизм (ГГ). Статеве дозрівання у чоловіків затримується. Описані атрофія та фіброз сім’явиносних канальців. При біопсії яєчок виявляють аплазію гермінативних клітин. Чоловіки з ГГ часто мають маленький пеніс, яєчка, що часто супроводжується гінекомастією. Рівень тестостерону та ФСГ (фолікулостимулюючого гормону) сироватки знижений.
Ендокринні порушення у жінок включають:
- зниження рівня гонадотропінів плазми;
- гірсутизм;
- порушення розвитку молочних залоз;
- полікістоз яєчників, передчасний статевий розвиток (пубертат у 8 років);
- ендометріоз;
- дис- чи аменорею.
Зовнішні статеві органи у жінок сформовані правильно. Пацієнти із синдромом Альстрема нефертильні.
Урологічні/ниркові порушення. Розлади сечовипускання спостерігаються у понад 50% хворих. Вони характеризуються диссинергією сфінктера уретри (відсутність координації сечового міхура та діяльності м’язового шару сечового каналу). Найбільш виражені ці прояви у дівчаток-підлітків. Незначні симптоми включають в себе – великі інтервали між вивільненням сечі, розлади сечовипускання. Помірні симптоми – нетримання сечі та симптоми, пов’язані з періодичним інфікуванням. Важкий перебіг супроводжується сечовими розладами, болями внизу живота, перитонеальним болем, який, зазвичай, пов’язаний з аномальною функцією міхура. Хронічна ниркова недостатність (ХНН) – пізній прояв хвороби. Першими ознаками ХНН є поліурія та полідипсія як результат порушення концентраційної функції нирок внаслідок фіброзних змін. При біопсії нирок часто виявляється фіброз, гломерулярний гіаліноз і атрофія канальців. Вік маніфестації та вираженість ХНН мінливі.
Печінкова дисфункція. Підвищена концентрація в плазмі крові трансаміназ частіше спостерігається у ранньому дитинстві. Печінкова недостатність може розвинутись у 20-30 років. При проведення біопсії виявляються різні ступені печінкового фіброзу, цирозу, постійного неспецифічного запалення із лімфоїдною інфільтрацією, вогнищевим некрозом та жировою дистрофією.
Легеневі зміни – утворення фіброзу та легеневої гіпертензії.
Панкреатит – дисліпідемія (гіпертригліцеридемія) – характерні для синдрому Альстрема. У хворих є ризик розвитку панкреатиту.
Інші прояви – сколіоз та кіфоз різного ступеня вираженості зустрічаються у третини хворих і носіїв синдрому Альстрема. Також можливий розвиток гіпотиреозу.
При підозрі на наявність у пацієнта синдрому Альстрема необхідно проведення наступного переліку досліджень:
- офтальмологічне обстеження із застосуванням електроретинографії та дослідженням полів зору;
- первинний запис антропометричних даних: зріст, вага, індекс маси тіла;
- аудіометрія (якщо пацієнт віком більше 4 років);
- консультація кардіолога з проведенням ЕКГ, Ехо-КГ;
- визначення рівню глюкози натще;
- після 6 років проведення тесту на толерантність до глюкози;
- визначення рівню інсуліну в плазмі крові;
- визначення рівню тригліцеридів крові натще, визначення рівню холестерину;
- уріналізіс та визначення рівню сечовини, сечової кислоти, креатинину та електролітів крові (контроль функції нирок);
- УЗД нирок та органів малого тазу;
- печінкові проби (АЛТ, АСТ) та УЗД печінки та жовчних шляхів;
- консультація невролога, ендокринолога.
Ускладнення:
З віком у хворих розвивається діабет 2 типу та інші порушення обміну речовин; ниркова недостатність. У багатьох пацієнтів формуються ускладнення з боку легенів та порушення функції печінки, з розвитком печінкової недостатності та портальної гіпертензії. У більшості чоловіків має місце гіпогонадотропний гіпогонадизм. Дилятаційна КМП призводить до виникнення серцевої недостатності та смерті.
Диференційний діагноз:
Синдром Барде-Бідля. Головні клінічні ознаки – пігментна дегенерація сітківки, дилятаційна полідактилія, ожиріння центрального генезу, розумова відсталість, гіпогонадизм, дисфункція нирок. Головна відмінність між цими синдромами полягає у дебюті захворювання. При синдромі Альстрема, зазвичай, візуальні прояви з’являються у перші 2 роки життя; а при синдромі Барде-Бідля – середній вік маніфестації 8,5 років. Полідактилія, яка є звичайним проявом при синдромі Барде-Бідля, не описана при синдромі Альстрема. Затримка розумового розвитку більш виражена при синдромі Барде-Бідля, ніж інтелектуальна дисфункція при синдромі Альстрема. Інші відмінності включають: відносно рідкісні порушення слуху (біля 5%), цукровий діабет (5-10%) при синдромі Барде Бідля порівняно з синдромом Альстрема. Тип успадкування аутосомно-рецесивний, описані мутації у щонайменше 8 генах.
Ахроматопсія. Патологія сітківки, що характеризується зниженою гостротою зору, ністагмом, світлобоязню, маленькою центральною скотомою, ексцентричною фіксацією, зниженням чи повною втратою кольорової чутливості. Більшість хворих мають повну ахроматопсію з відсутністю функції усіх трьох видів паличок, тобто чутливих до довгих хвиль – червоний спектр; хвиль середньої довжини – зелений; і до хвиль короткої довжини – блакитний. Рідко пацієнти мають часткову ахроматопсію, при якій один чи більше типів паличок частково функціонують, що проявляється менш тяжким перебігом хвороби. Ністагм і підвищена чутливість до яскравого світла розвиваються на початку захворювання. Гострота зору знижується через деякий час. Очне дно без патології. Визначаються мутації у 3 генах: CNGA3, CNGB3, GNAT2. Тип успадкування аутосомно-рецесивнийх.
Вроджений амавроз Лебера (LCA). Тяжка дегенерація сітківки без ураження інших органів та систем стає очевидною на першому році життя. Зниження зору супроводжується ністагмом, світлобоязню та гіперметропією. Зміни на ЕРГ не характерні. Сітківка до 1 року нормальна. Пігментна ретинопатія – схильність до накопичення пігменту сітківкою – частіше з’являється у більш пізніші строки. В даний час відомо 6 генів пов’язаних з LCA – CRX, GRB1, GUCY2D, AIIPL1, RPGRIP1 та RPC65, мутації в яких вважаються причиною розвитку амаврозу Лебера в 1/3-1/2 частині випадків. Частіше за все LCA успадковується аутосомно-рецесивно (АР), рідше аутосомно-домінантно (АД) – як результат мутації CRX.
Рання дилятаційна КМП. Характеризується розширенням меж серця і зниженням систолічної функції – це кінцева стадія низки спадкових та набутих хвороб. Родинна дилятаційна КМП успадковується АД, рідше АР – з вентрикулярною дилятацією і систолічною дисфункцією – яка маніфестує у 30-40 років.
Спадкові мітохондріальні хвороби. Це група хвороб, виникнення яких викликане мутаціями у мітохондріальній чи ядерній ДНК. Клінічні прояви, спільні із синдромом Альстрема:
- кардіоміопатія;
- нейросенсорна глухота;
- атрофія зорового нерву;
- пігментна ретинопатія;
- цукровий діабет.
Однак клінічні прояви мітохондріальних хвороб включають ще м’язову гіпотонію та порушення функції ЦНС, ці ознаки є нетиповими при синдромі Альстрема. Загалом, ці хвороби маніфестують у пізньому дитинстві чи у дорослому віці, на відміну від синдрому Альстрема, який проявляється протягом першого року життя.
Лікування:
Специфічного лікування не розроблено. Терапія симптоматична:
- При фотодисфорії – використання кольорових лінз.
- При ожирінні – лікувальне харчування за загальними принципами, регулярне фізичне навантаження.
- При нейросенсорній глухоті – хірургічна корекція у пацієнтів з ексудативним отитом. Цифрові слухові апарати приносять полегшення деяким хворим.
- При дилятаційній КМП – інгібітори АПФ, сечогінні; при розвитку СН – серцеві глікозиди, ß-адреноблокатори.
- При цукровому діабеті – лікування за загальними принципами.
- При гіпертригліцеридемії – високі дози статинів, нікотинова кислота; фібрати в деяких випадках неефективні.
- При урологічних порушеннях – деяким хворим потрібен сечовий катетер, щоб уникнути труднощів з сечовиділенням. Якщо є протеїнурія використовують інгібітори АПФ.
- При портальній гіпертензії – ß-адреноблокатори та склерозування езофагальних вен.
- При хронічній обструктивній хворобі легень – лікування відповідно до загально прийнятих протоколів.
- При гіпотиреозі – терапія тироксином.
Приблизний план диспансеризації:
- Дегенерація сітківки – щорічний огляд офтальмолога, перевірка полів зору, ЕРГ.
- Ожиріння – вимірювання ваги, росту та визначення ІМТ (індекс маси тіла) кожен рік графік кривої приросту.
- Прогресуюча нейросенсорна глухота – аудіометрія (рекомендована після 4-х років) для виявлення нейросенсорної та провідної глухоти.
- Дилятаційна КМП – детальний анамнез, огляд кардіолога (щорічно), ЕхоКГ, ЕКГ з 5 років, пізніше кожні 2 роки (при наявності ознак серцевої дисфункції – пітливість, втомлюваність, летаргія, акроціаноз, зниження фізичної активності), при потребі запис ЕКГ протягом 24 годин – Холтерівський моніторинг.
- Цукровий діабет 2 типу – натщесерце визначають концентрацію глюкози у плазмі кожні 2-3 місяці навіть у дітей 1 року життя. Якщо натщесерце рівень глюкози перевищує 7 ммоль/л чи післяобідня глюкоза більша ніж 11 ммоль/л, то потрібне регулярне вимірювання HbA1 та концентрації глюкози у сироватці (кожні 6 місяців, а можливо і частіший діабетичний контроль). Визначення толерантності до глюкози з 6 років, визначення інсуліну в плазмі крові щорічно (гіперінсулінемія можлива і у дітей до року).
- Гіпертригліцеридемія: визначається ліпідний профіль кожен рік.
- Гіпогонадотропний гіпогонадизм – огляд ендокринолога, визначення рівня статевих гормонів.
- Урологічна/ниркова дисфункція: урінолізіс, визначення концентрації сечовини, електролітів, сечової кислоти та креатинину плазми двічі на рік; УЗД нирок, сечового міхура – 1-2 рази на рік.
- Печінкова дисфункція – вимірювання аланінамінотрансферази; УЗД щорічно для виявлення гепатомегалії та портальної гіпертензії.
- Легенева дисфункція – при наявності клінічних симптомів: проведення спірометрії, функціональних тестів.
- Профілактика виникнення панкреатиту: визначення рівню тригліцеридів сироватки та концентрації холестерину – щорічно планово та в період будь-якого захворювання та дегідратації.
Тип успадкування:
Аутосомно-рецесивний. Батьки пробанда є облігатними гетерозиготами, вони таким чином несуть кожен один мутантний алель. Ризик для сибсів пробанду мати хворобу Альстрема складає 25%, ризик гетерозиготного носійства для сибсів 50%.
В родині необхідно проведення преконцепційної профілактики та пренатальної діагностики з застосуванням молекулярної діагностики при кожній наступній вагітності, враховуючи 25% ризик для сибсів.
Номер з каталогу МІМ:
203800 Alstrom Syndrome; ALMS
606844 ALMS1 Gene; ALMS1
Література:
- Bronson R. Pathological findings in Alstrom syndrome. Alstrom Syndrome International Conference, Ottawa. 2001.
- Collin GB, Marshall JD, Boerkoel CF, Levin AV, Weksberg R, Greenberg J, Michaud JL, Naggert JK, Nishina PM. Alstrom syndrome: further evidence for linkage to human chromosome 2p13. Hum Genet 1999;105:474-9.
- COPD Guideline Group of the Standards of Care Committee of the BTS. BTS guidelines for the management of chronic obstructive pulmonary disease. Thorax 1997;52(Suppl 5):1-28.
- Deeble VJ, Roberts E, Jackson A, Lench N, Karbani G, Woods CG. The continuing failure to recognise Alstrom syndrome and further evidence of genetic homogeneity. J Med Genet 2000;37:219.
- Hearn T, Renforth GL, Spalluto C, Hanley NA, Piper K, Brickwood S, White C, Connolly V, Taylor JF, Russell-Eggitt I, Bonneau D, Walker M, Wilson DI. Mutation of ALMS1, a large gene with a tandem repeat encoding 47 amino acids, causes Alstrom syndrome. Nat Genet 2002;31:79-83.
- Maffei P, Munno V, Marshall JD, Scandellari C, Sicolo N. The Alstrom syndrome: is it a rare or unknown disease? Ann Ital Med Int 2002;17:221-8.
- Marshall JD, Ludman MD, Shea SE, Salisbury SR, Willi SM, LaRoche RG, Nishina PM. Genealogy, natural history, and phenotype of Alstrom syndrome in a large Acadian kindred and three additional families. Am J Med Genet 1997;73:150-61.
- Marshall JD, et al. New Alstrom Syndrome phenotypes based on the evaluation of 182 cases. Arch Intern Med 2005;165:675-683.
- Michaud JL, Heon E, Guilbert F, Weill J, Puech B, Benson L, Smallhorn JF, Shuman CT, Buncic JR, Levin AV, Weksberg R, Breviere GM. Natural history of Alstrom syndrome in early childhood: onset with dilated cardiomyopathy. J Pediatr 1996;128:225-9.
- Paisey RB, Carey CM, Parkinson MJ, Parkinson C, Cole MD. Alstrom syndrome-the case for secondary prevention. Diabet Res Clin Pr 2000;50(suppl 1):202.
- Russell-Eggitt IM, Clayton PT, Coffey R, Kriss A, Taylor DS, Taylor JF. Alstrom syndrome. Report of 22 cases and literature review. Ophthalmology 1998;105:1274-80.
- Titomanlio L, De Brasi D, Buoninconti A, Sperandeo MP, Pepe A, Andria G, Sebastio G. Alstrom syndrome: intrafamilial phenotypic variability in sibs with a novel nonsense mutation of the ALMS1 gene. Clin Genet 2004;65:156-7.
- Van den Abeele K, Craen M, Schuil J, Meire FM. Ophthalmologic and systemic features of the Alstrom syndrome: report of 9 cases. Bull Soc Belge Opthalmol 2001;281:67-72.
Переглянуто редакційною колегією I.B.I.S.: 26/05/2005
Дивіться також:
- Синдром Альстрема (інформація для батьків)
Вживання алкоголю під час вагітності
(Alcogol Use During Pregnancy)
Координатор Українського Альянсу
із запобігання вродженим вадам розвитку
Вживання алкоголю під час вагітності може призвести до виникнення фізичних вад розвитку та розумової затримки. Кожного року більше 50 тисяч немовлят народжується з різними проявами уражень, що пов’язані з вживанням алкоголю. Однак багато жінок, які усвідомлюють, що вживання спиртних напоїв у значній кількості під час вагітності може призвести до виникнення вроджених вад, не уявляють собі, що помірне чи навіть випадкове вживання спиртних напоїв також завдає шкоди плоду.
До речі, не доведено існування такого рівня алкоголю, щоб був нешкідливим під час вагітності. Тому вагітним жінкам рекомендують утриматися від вживання алкоголю, включно пива, вина, шампанського, прохолодні напоїв, що містять алкоголь та лікери під час вагітності та всього періоду годування грудьми. До того ж, часто жінки протягом перших тижнів не знають, що вони вагітні. Жінки, які планують вагітність, повинні відмовитися від алкогольних напоїв.
Недавні дослідження показали, що жінки, які очікують на вагітність, і продовжують вживати алкоголь навіть у малих кількостях, можуть знизити свою можливість завагітніти.
Як відмічається у нещодавньому державному соціологічному опитуванні, у період між 1991 та 1995 роками відмічалося зростання вживання алкоголю вагітними жінками. У 1995 році кількість жінок, які часто вживали алкоголь, була в чотири рази більшою ніж у 1991 році. Частим вважається вживання алкоголю невеликими порціями кожного дня або близько п’яти порцій за один день у попередньому місяці. В опитуванні говориться, що у 1995 році приблизно 140 тисяч вагітних жінок (що становить десь 3,5%) вживали алкоголь постійно у порівнянні з 1991 роком коли відмічалось 32 тисячі жінок, що становило менше 1%. Жінки, які зловживають алкоголем, в багато разів збільшують ризик розвитку у своїх дітей алкоголь-залежних уражень. В опитуванні також говоритися, що у 1995 році кількість вагітних жінок, які хоч один раз вживали алкоголь попереднього місяця, становила 16% у порівнянні з 12% у 1991 році.
Коли вагітна жінка вживає алкоголь, він без затримки проходить через плаценту безпосередньо до плода. В незрілому організмі ненародженої дитини алкоголь розкладається набагато повільніше, ніж у дорослої людини. В результаті чого рівень алкоголю в крові плода може бути навіть вищим і лишатися у підвищеному стані довше, ніж в крові матері, що може призвести до важких незворотніх змін різних органів та систем ще ненародженої дитини.
Чим небезпечне вживання алкоголю під час вагітності?
За даними Інституту Медицини кожного року в США народжується від 2 до 12 тисяч дітей з Алкогольним синдромом плода (FAS) – поєднанням фізичних та розумових вад розвитку. До 40% дітей з Алкогольним синдромом плода народжується у жінок хворих на алкоголізм. Це жінки які постійно вживали алкоголь на протязі всієї вагітності або мали часто повторюванні випивки (п’ять чи більше порцій за один раз).
Алкогольний синдром плода – найбільш поширена з відомих причин, що викликають розумову затримку і єдина причина, якої можна уникнути. Діти з класичним алкогольним синдромом плода народжуються із затримкою розвитку, і як правило з віком погано набирають вагу. Такі діти мають характерний вигляд: короткі очні щілини, короткий кирпатий ніс, збільшену відстань між носом і верхньою губою. Органи у них не формуються належним чином, особливо серце. У багатьох дітей з алкогольним синдромом плода головний мозок малий за розміром і ненормально сформований, більшість мають той чи інший ступінь розумової відсталості. У багатьох відмічається погана координація рухів і неможливість зосереджуватись, також існують складнощі у поведінці.
Дія алкогольного синдрому плода триває протягом всього життя. Навіть, коли немає розумової відсталості, підлітки та дорослі з діагнозом “алкогольний синдром плода” мають різного ступеню психологічні та поведінкові труднощі, що часто ускладнює їм виконання роботи і можливість жити без сторонньої допомоги.
В десять разів більше дітей народжується з неповним алкогольним синдромом плода, що мають не всі прояви алкоголь-залежних уражень. Цей стан деколи називають “Дія алкоголю на плід” (Fetal Аlcohol Effects – FAE). У таких дітей можуть бути деякі з фізичних чи розумових вроджених вад, що пов’язані з алкогольним синдром плода. Нещодавно Інститут Медицини запропонував нову особливу діагностичну категорію для “Дії алкоголю на плід” (FAE), відносити фізичні вроджені вади розвитку (такі як вади серця) до алкоголь-залежних вроджених вад, а затримки психічного розвитку та вади поведінки розцінювати як алкоголь-залежні розлади нервової системи.
Яка кількість алкоголю є надмірною під час вагітності?
Не доведено, що існує нешкідливий рівень алкоголю в крові матері під час вагітності. Повна картина Алкогольного синдрому плода як правило проявляється у нащадків хронічних алкоголіків, дуже часто у жінок, які вживають по чотири, п’ять чи більше порцій алкоголю на день. Однак, це трапляється і у жінок, які п’ють меньше. “Дія алкоголю на плід” (FAE) може виникнути у дітей, народжених жінками, які під час вагітності вживають легкі алкогольні напої і не так часто.
Порівняно з дітьми з алкогольним синдромом плода, менш відомі віддалені наслідки для дітей з FAE. Дослідження Др. Рональда Броуна та групи вчених з Emory University, Атланта, який спостерігав до 10-річного віку за групою дітей, що зазнали впливу алкоголю до народження, але не мали повного прояву алкогольного синдрому плода, показали, що коли ці діти виростають до шкільного віку, вони не тільки показують більш низькі показники інтелектуальних можливостей, але і проявляють гіперактивність (як це називають педагоги): агресивність, схильність до руйнування, неуважність та нервозність. В інших дослідженнях дітей шкільного віку, що зазнали впливу алкоголю, показано, що наряду з проблемами поведінки, у них існують труднощі з навчанням, особливо з математикою та запам’ятовуванням.
Вчені пильно ставляться до незначної дії помірного вживання спиртних напоїв або вживання більш легких алкогольних напоїв під час вагітності. У дослідженнях, що були проведені в університетах Вашингтону та Сіетла, спостерігали за групою дітей шкільного віку (до 14 років), чиї матері вживали під час вагітності три або більше порцій алкогольних напоїв на день. При проходженні тестів на розумові здібності у віці 4-х років, ці діти показали рівень на п’ять пунктів нижчий за середній показник дітей контрольної групи. Так само дослідження, проведене у Франції, 1995 року, показало, що тести 4,5-річних дітей, народжених від матерів, які вживали принаймі три порції алкогольних напоїв на день, на сім пунктів нижчі за показникі інших дітей. Вчені з Сіетла також виявили зростаючу імовірність проблем у навчанні (включно проблеми з математикою) у 7-ми річному та 14-ти річному віці у дітей народжених від помірно вживаючих алкоголь матерів.
Якщо вагітна жінка прийняла одну чи дві порції алкогольних напоїв ще до того, як вона зрозуміла, що є вагітною, чи може це зашкодити плоду?
Недоведено, що плід може зазнати шкоди від прийняття вагітною жінкою однієї чи двох порцій алкогольних напоїв ще до того, як вона зрозуміла, що є вагітною. Звичайно розвиток головного мозку плода та інших органів розпочинається на третьому тижні вагітності і є легко уразливим протягом перших трьох тижнів. Оскільки не існує нешкідливих доз алкоголю, жінка повинна припинити вживання алкоголю в той момент, коли вона тільки запідозрить, що може бути вагітною і утримуватись від вживання всіх видів алкогольних напоїв якщо вона очікує на вагітність.
Які ще проблеми пов’язані з вживанням алкоголю під час вагітності?
Вживання алкоголю під час вагітності підвищує ризик викиднів, народжень дітей з низькою вагою, мертвонароджень та смерті дитини у періоді ранньої новонародженості. Ті, хто зловживають алкоголем, мають у 2-4 рази вищий ризик мати викидні у 4-6 місяців вагітності ніж ті, хто не вживає алкоголь. Вони також можуть вдвічі частіше втрачати дитину у перінатальний період – з 28-ого тижня вагітності до перших тижнів після народження.
Чи небезпечне вживання алкоголю під час годування грудьми?
Алкоголь навіть у маленькій кількості потрапляє у грудне молоко і потім в організм дитини. В одному з досліджень було показано, що у дітей, які знаходились на природньому (грудному) годуванні жінками, що вживали одну-дві порції алкоголю на день, більш повільно розвивались рухові навички (такі як повзання та ходіння) у порівнянні з дітьми, які не зазнали впливу алкоголю. Більші дози алкоголю також можуть потрапляти через грудне молоко при годуванні. Тому всім жінкам рекомендують утримуватись від вживання алкоголю на весь період годування грудьми.
Чи може вживання батьком алкоголю у великій кількості призвести до виникнення алкогольного синдрому плода (FAS)?
На сьогодні немає доказів того, що вживання алкоголю у великій кількості батьком може призвести до виникнення алкогольного синдрому плода. Але зростає кількість даних про те, що вживання алкоголю у великій кількості чоловіком може мати негативний вплив на перебіг вагітності та здоров’я дитини. Вживання алкоголю у великій кількості чоловіком може знизити рівень чоловічого гормону тестостерону, що призводить до зменшення кількості сперми, а іноді і до безпліддя.
Щоб повністю з’ясувати, яким чином вживання чоловіком алкоголю може вплинути на результат вагітності, потрібно більше досліджень. Чоловік, який припинить вживати алкоголь на час вагітності своєї партнерки, також допоможе їй відмовитись від алкоголю.
Переглянуто редакційною колегією I.B.I.S.: 15/02/2002
Дивіться також:
- Фетальний алкогольний синдром
- Фетальний алкогольний синдром (ФАС) – одна із найпоширеніших причин вроджених вад і порушень розвитку дітей (інформація для батьків)
- Алкогольний синдром плода (інформація для батьків)
- Алкоголь та здоров’я (інформація для батьків)
|
|
|
|
|
|




