
Uncategorized
Фолієва кислота – спеціальне видання

Червень, 2000 р.
Усі матеріали випуску, за винятком підписаних, надані проф. В. Вертелецьким (США).
Зміст

- Фолієва кислота: справжній переворот у медицині можливий
- Вроджені вади розвитку – одна з головних причин дитячої смертності та інвалідності в усьому світі
- Spina bifida (інформація для лікарів)
- Спинно-мозкова кила (інформація для населення)
- Аненцефалія (інформація для лікарів)
- Фолієва кислота – роль у запобіганні вадам невральної трубки (інформація для медичних працівників)
- Фолієва кислота і рак товстої кишки
- Фолієва кислота і атеросклероз
- Ефективність профілактики фолієвою кислотою вад невральної трубки
- Фолієва кислота (інформація для населення)
- Показники частоти вроджених дефектів та вад розвитку серед новонароджених США (дані 1997 року)
- Графік зростання обізнаності жінок дітородного віку в США про корисність фолієвої кислоти з 1995 по 1997 рр.
- Деякі продукти – джерела фолатів
- Рецепти страв з високим вмістом фолатів
- Роль фолієвої кислоти в запобіганні вадам невральної трубки: огляд літератури
- Фолієва кислота: бібліографія
- Інформація про фолієву кислоту в IBIS
Спеціальне видання “Гено-Терато-Епі-Том” висвітлює основні факти про останні наукові досягнення і клінічні спостереження, що мають відношення до профілактики і зменшення частоти вроджених аномалій і порушення розумового розвитку у дітей.
“Гено-Терато-Епі-Том” – це інформаційне видання, редактором якого є професор Володимир Вертелецький.
Ми вітаємо Ваш внесок у “Гено-Терато-Епі-Том” у вигляді коментарів, матеріалів, перекладів на інші мови тощо.
Листи англійською мовою посилати проф. В. Вертелецькому.
Спеціальне видання “Гено-Терато-Епі-Том”
Випуски:
- Талідомід – спеціальний випуск англійською мовою (1999 рік)
- Вроджені вади – загальний огляд англійською мовою (грудень 1999 року)
- Талідомід – спеціальний випуск (березень 2000 року)
- Фолієва кислота – спеціальний випуск (червень 2000 року)
- Новини – спеціальний випуск англійською мовою (липень 2006 року)
Спеціальне видання “Гено-Терато-Епі-Том” висвітлює основні факти про останні наукові досягнення і клінічні спостереження, що мають відношення до профілактики і зменшення частоти вроджених аномалій і порушення розумового розвитку у дітей.
“Гено-Терато-Епі-Том” – це інформаційне видання, редактором якого є професор Володимир Вертелецький.
Ми вітаємо Ваш внесок у “Гено-Терато-Епі-Том” у вигляді коментарів, матеріалів, перекладів на інші мови тощо.
Листи англійською мовою посилати проф. В. Вертелецькому.
Краніосиностоз
(Craniosynostosis)
В.В. Гуштаб
Неонатолог відділення патології новонароджених
Рівненської обласної дитячої лікарні
Краніосиностоз – вроджена вада розвитку кісток черепа, яка характеризується передчасним зрощенням одного, декількох або всіх черепних швів, що призводить до деформації черепа і часто вторинних неврологічних порушень.
Захворювання вперше описав в 1951 Вірхов.
Діагностика:
Діагностика краніосиностозу грунтується на клінічних і рентгенологічних даних. Мінімальною діагностичною ознакою є аномальна форма черепа. Прояви захворювання залежать від часу виникнення, поширеності та вираженості синостозу (зрощення) черепних швів. Описані різні форми деформації черепа: баштовий (акроцефалія), ладдєподібний (скафоцефалія), трикутний (тригоноцефалія), косий (плагіоцефалія), уступоподібний (батроцефалія), плоский (платицефалія), вузький (лептолія), гострокінцевий (цукрова голова) та інші. Своєрідними проявами краніосиностозу є синдром Аперта, черепно-лицевий дизостоз (синдром Крузона). Ще виділяють компенсовану і декомпенсовану форму краніосиностозу. Перша проявляється незначними явищами декомпенсації у вигляді періодичного головного болю. Декомпенсована форма характеризується приступами головного болю, нудотою, блювотою, застійними явищами на очному дні або атрофією дисків зорових нервів (первинна або вторинна), може бути асиметрія сухожильно-надкісткових рефлексів, судоми, затримка психо-моторного розвитку. У хворих часто зустрічається екзофтальм, у виникненні якого, крім механічних факторів, важливу роль відіграє застій в очних венах та їх розширення. Для краніосиностозу характерні своєрідні зміни на краніограмах: витончення кісток склепіння черепа і посилення пальцевих вдавлень (симптом “кованого срібла”), деформація основи, поглиблення турецького сідла, черепних ямок, борозен і синусів, затримка пневматизації лобної та тім’яної кісток, розходження швів, які “не встигли” облітеруватися. Спинно-мозкова рідина при люмбальній пункції витікає під підвищеним тиском, цитоз і кількість білка в ній не змінені.
Наслідком краніосиностозу є деформації черепа та підвищення внутрішньочерепного тиску.
Частіше всього зустрічається скафоцефалія (довгий вузький череп з виступаючим лобом і потилицею) внаслідок передчасного заростання стрілоподібного шва. Туррицефалія з’являється через передчасне заростання коронарного і стрілоподібного швів. Брахіцефалія – внаслідок зрощення коронарного шва.
Диференційний діагноз:
Краніосиностоз потрібно диференціювати з мікроцефалією, черепно-лицевим дизостозом, акроцефалосиндактилією, кефалогематомою, внутрішньочерепною пухлиною, спадковою доброякісною макроцефалією. При підозрі на патологію магістральних судин проводиться паротидна і вертебральна ангіографія. Як тільки виявлено краніосиностоз, потрібно бути досить обережним у діагностуванні, щоб не сплутати з потиличною плагіоцефалією, так як позиційні зміни можуть бути схожими на цю дуже рідкісну форму краніосиностозу.
Етіологія і патогенез:
Етіологія і патогенез краніосиностозу до кінця не встановлені. Вважають, що етіологічним фактором краніосиностозу є патологія фактора росту фібробластів чи його рецепторів. Мають значення різні спадкові і внутрішньоутробні порушення. Існує припущення про первинні зміни, які з’являються в самому черепі, що можуть призвести до передчасного окостеніння швів, зменшення об’єму внутрішньочерепної порожнини при нормальному рості мозку. Встановлена роль первинної патології розвитку судинної системи голови (екстра- та інтракраніальні судини). Це може призвести до венозного застою з підвищенням внутрішньочерепного тиску і неправильного розвитку магістральних артерій голови.
Для опису краніосиностозу зазвичай використовуються дві класифікації:
- Проста (залучений лише один шов) чи комплексна (залучено більше одного шва).
- Первинна (спричинена ідіопатичним ненормальним злиттям кісток) чи вторинна (спричинена різними системними чи неврологічними захворюваннями).
Популяційна частота:
Основний критерій – передчасне закриття одного чи більше краніальних швів – проявляється у одного з 2000-2500 новонароджених.
Тип успадкування: aутосомно-домінантний і аутосомно-рецесивний.
Лікування:
При субкомпенсованих формах призначають дегідратуючу терапію (діакарб або гліцерин, рідше – лазікс). Дозування препаратів і тривалість прийому ті ж, що і при гідроцефалії. Декомпенсовані форми з вираженою внутрішньочерепною гіпертензією, зниженням зору, розвитком судинної енцефалопатії є показами до оперативного втручання. Проводиться двобічна шматкова бокова краніотомія з утворенням вільних або фіксованих кісткових шматків.
Номер з каталогу МІМ:
123100 Craniosynostosis, Type 1; CRS1
Література:
- Warkany J. Congenital Malformations: Notes and Comments. Year Book Publisher. Chicago, 1981:893.
- Козлова С.И. и др. Наследственные синдромы и медико-генетическое консультирование. Справочник. Л.: Медицина, 1987.- C. 96.
- Ромазанов А.П., Лященко Д.С. Роль патологии сосудов в развитии краниостеноза.- С. 62-73.
- Энциклопедия детского невролога /Под ред. Г.Г. Шанько.- Минск: Беларусская энциклопедия, 1993.- C. 169-170.
Переглянуто редакційною колегією I.B.I.S.: 4/01/2002
Нунан синдром
(Noonan Syndrome)
Леся Юріївна Височина
Лікар акушер-гінеколог поліклініки № 3,
м. Рівне
Включення:
- Синдром Нунан;
- Чоловічий синдром Тернера;
- Жіночий синдром Тернера з нормальним ХХ каріотипом;
- Жіночий псевдотернер синдром;
- Тернеровський фенотип з нормальним каріотипом;
- Синдром Ульріха;
- Синдром Ульріха-Нунан.
Виключення:
- Синдром Кліпеля-Фейля;
- Синдром Тернера;
- Синдром Аарського;
- Синдром множинних лентиго.
Основні діагностичні критерії:
Крилоподібні складки на шиї, аномалії грудної клітки, крипторхізм, вроджені вади правих відділів серця.
Клінічна картина:
Типові риси обличчя: гіпертелоризм, епікант, антимонголоїдний розріз очей, птоз повік, мікрогнатія. Вуха низько розміщені зі складчастим завитком, аркоподібне піднебіння, розщілина язичка, порушення прикусу, міопія, кератоконус, косоокість, низький ріст волосся на потилиці, крилоподібні складки на шиї. Шия коротка, широка. Грудна клітка щитоподібна, гіпертелоризм сосків. Характерний низький зріст, вальгусна деформація ліктьових суглобів, іноді кіфосколіоз. Можливий периферичний лімфатичний набряк. В 55% наявні ВВС і великих судин, причому при 80% випадків уражені праві відділи серця. У 27% відмічаються вади сечовивідної системи. Характерний гірсутизм. Функція статевих залоз може бути збереженою або відсутньою. Розумова відсталість (дебільність) виражена у 61% хворих, але є пацієнти з нормальним або високим інтелектом.
Асоційовані вади:
Колобома, гіпоплазія сосків, шийні ребра, набряк тилу стопи і кисті, хілоторакс, імунологічна водянка, гепатоспленомегалія, шкірні невуси, гіпереластичність шкіри, злоякісна гіпертермія, ювенільна мієломоноцитарна лейкемія.
Диференційний діагноз:
Синдром Кліпеля-Фейля, синдром Тернера, синдром Аарского, синдром множинних лентиго, синдром Вільямса.
Етіологія:
Аутосомно-домінантний тип успадкування.
Ризик для сибсів: 50%.
Ризик для потомства: 50%.
Частота виникнення: може досягати 1 на 1000 живонароджених.
Співвідношення між статями: Ч1 : Ж1.
Патогенез:
Первинний дефект не виявлено. Характерне ураження багатьох систем.
Вік прояву:
У періоді новонародженості, якщо є лімфедема або крилоподібні складки на шиї. В подальшому увагу привертають низький зріст, характерні риси обличчя, ураження органів і систем.
Прогноз:
Тривалість життя може бути нормальна, зменшується при ВВС і їх ускладненні. Розумова відсталість виражена у 61% хворих. Приєднання скелетних аномалій може знижувати працездатність. Особливу увагу варто приділяти працевлаштуванню: вибір спеціальності повинен сприяти соціальній адаптації хворих з урахуванням їхніх розумових здібностей і фізичних недоліків.
Ускладнення:
Ураження серцево-судинної системи, безпліддя, більша частота розвитку новоутворів, порівняно зі здоровою популяцією.
Профілактика: медико-генетичне консультування.
Лікування:
Симптоматичне. За наявності ознак гіпогонадизму лікування полягає в замісній терапії препаратами статевих гормонів. Застосування гормону росту неефективне. За показами проводиться хірургічна корекція вроджених вад розвитку, лікування психічних порушень, пов’язаних із затримкою розумового розвитку.
Номер з каталогу МІМ:
163950 Noonan Syndrome 1; NS1
Література:
- Бабцева А.Ф., Климова Н.В., Юткина О.С., Лабзин В.И., Ермаков Г.А. Медицинская генетика: Учебно-методическое пособие для студентов, врачей-интернов, ординаторов, педиатров.- Благовещенск: Амурская государственная медицинская академия, 2002.– с. 58.
- Козлова С.И., Демикова Н.С., Семанова Е., Блинникова О.Е. Наследственные синдромы и медико–генетическое консультирование.- М.: Практика, 1996.- C.192-193.
- Birth Defects Compendium. D. Bergsma (ed.). Second Edition. New York, 1979:778-779.
- Gorlin RJ, Cohen MM, Hennekam RCM. Syndromes of the Head and the Neck. 4th edition. Oxford University Press. 2001:1000-1002.
- Jones KL. Smith’s Recognizable Patterns of Human Malformations. 6th edition. W.B. Sauders Company. Philadelphia. 2002:124.
- Noonan Syndrome. Manbir Online: Diseases and Conditions.
Переглянуто редакційною колегією I.B.I.S.: 14/10/2010
Дивіться також:
- Синдром Нунан – зріст хлопчиків (діаграма розвитку)
- Синдром Нунан – зріст дівчат (діаграма розвитку)
Синдром Елерса-Данлоса
(Ehlers Danlos Syndrome)
Г.В. Скибан
Зав. поліклінікою науково-дослідного інституту
педіатрії, акушерства і гінекології (НДІ ПАГ)
м. Київ
Основні діагностичні критерії:
Гіпереластичність і крихкість шкіри, її витонченість типу “цигаркового паперу”, гіперрухомість суглобів, підвищена кровоточивість.
Клінічна характеристика:
Вперше описаний голандським дослідником Van Meckeren’ом. Російський дерматолог А.Н.Черногубов в 1892 році вперше описав захворювання як генералізований дефект сполучної тканини в ембріональному періоді. У радянській літературі цей синдром іноді визначається як синдром Черногубова.
Клінічне синдром являє собою групу захворювань сполучної тканини, які включають дефекти шкіри і суглобів і різняться за типом успадкування, клінічними особливостями та біохімічним дефектом.
І тип (важка форма) характеризується генералізованістю і тяжкою гіперрухомістю суглобів, вираженою гіперпластичністю шкіри. Ранимість шкіри призводить до утворень “папіросних” келоїдних рубців. В ділянці ліктів та колін часто мають місце підшкірні припухлості, а на передній поверхні гомілок – підшкірні вузлики. Характерні варикозні розширення вен. Гіперрухомість суглобів викликає ортопедичні проблеми. У новонароджених може бути виражена м’язова слабкість. Рідше захворювання включає голубі склери та зміщення кришталика, а також діафрагмальну килу, дивертикул кишківника або сечового міхура, мегаезофагус, мегатрахею, мегаколокабо невризму аорти, пролапс мітрального клапана).
При II типі (середньої важкості) гіперрухомість суглобів обмежується кистями і ступнями. Шкірні дефекти незначні. Може бути пролапс мітрального клапана.
І і II типи характеризуються підвищеною розчинністю колагену шкіри, зміненою архітектури колагенових волокон: їх діаметр більший, ніж у нормі.
III тип (тип сімейної гіпермобільності суглобів) включає виражену гіпермобільність усіх суглобів без м’язово – скелетних деформацій. Зміни шкіри практично відсутні.
IV тип (артеріальний, екхіматозний, типу Sack’а) – найважча форма, оскільки при ній можливі смертельні випадки внаслідок спонтанних розривів великих судин і перфорації кишечника. Шкіра надзвичайно витончена, не розтягується, через неї просвічуються вени. Описана знижена кількість колагену. Superti – Furga et al. (1988) описали мутацію гена COL 3A1.
V тип (Х-зчеплений) характеризується мінімальною гіперрухомістю суглобів і різко вираженою гіпереластичністю шкіри, її ламкістю, утворенням підшкірних пухлин і вузликів, генералізованою м’язово-скелетною слабкістю (дорзальний кифоз, кила). Клінічно нагадує тип II. Біохімічне виявляється недостатністю активності лізилоксидази, ферменту, який бере участь у синтезі колагену.
VI тип (очний) характеризується важким сколіозом, помірним ураженням шкіри і суглобів, ламкістю тканин ока. Невеликі травми спричиняють розрив склери і рогівки, відшарування сітківки. Біохімічне розрізняють три підтипи:
- А – важка форма – відсутність гідроксилізину в колагені шкіри і низька активність лізилгідроксилази;
- В – клінічно подібна – нормальний рівень гідроксилізину і низька активність лізилгідроксилази в фібробластах шкіри;
- С – принципово очна форма – нормальні біохімічні показники.
Тип VII включає низький ріст, генералізовану гіперрухомість суглобів, підвивихи кульшових, колінних, ліктьових і гомілково-ступеневих суглобів, сколіоз. Шкіра млява, помірно гіпереластична і ламка. Для обличчя характерні епікант, гіпертелоризм, вдавлене перенісся, мікрогнатія. Подібно до типу І біохімічне визначаються порушення обміну колагену. В залежності від наявності дефекту про альфа 1 чи про альфа 2 ланцюга пропептиду визначають два підтипи.
Тип VIII (періодонтальний) характеризується періодонтозом, ранньою втратою зубів, відсутністю альвеолярних кісток, вираженими гіперрухомістю суглобів та гіпереластичністю і ламкістю шкіри.
Тип Х (дефект фібронектину) представляє гіперрухомість суглобів, гіпереластичність і ламкість шкіри, дрібні крововиливи, проте без масивної кровотечі. Біохімічно: дефіцит фібронектину.
Ускладнення:
Пов’язані з ортопедичними деформаціями, патологією серця.
Профілактика:
Рекомендоване медико-генетичне консультування.
Популяційна частота: 1: 100 000.
Тип успадкування:
Аутосомно-домінантний для І, ІІ, ІІІ типів; аутосомно-рецесивний – для IV, VII типів; Х-зчеплений рецесивний для V типу; домінантний і рецесивний для IV типу; очевидно аутосомно-рецесивний для VIII типу.
Диференційний діагноз:
Синдром Марфана, незавершений остеогенез, синдром Дауна, синдром Ларсена.
Номер з каталогу МІМ:
130000 Ehlers-Danlos Syndrome, Type I
130010 Ehlers-Danlos Syndrome, Type II
130020 Ehlers-Danlos Syndrome, Type III
130050 Ehlers-Danlos Syndrome, Type IV, Autosomal Dominant
305200 Ehlers-Danlos Syndrome, Type V
225400 Ehlers-Danlos Syndrome, Type VI
130060 Ehlers-Danlos Syndrome, Type VII, Autosomal Dominant
225410 Ehlers-Danlos Syndrome, Type VII, Autosomal Recessive
130080 Ehlers-Danlos Syndrome, Type VIII
130070 Ehlers-Danlos Syndrome, Progeroid Form
130090 Ehlers-Danlos Syndrome, Autosomal Dominant, Type Unspecified
225310 Ehlers-Danlos Syndrome with Platelet Dysfunction from Fibronectin Abnormality
225320 Ehlers-Danlos Syndrome, Autosomal Recessive, Cardiac Valvular Form
Літературні джерела:
- Buyse M.L. Birth Defects Encyclopedia: Center for Birth Defects Information Services, Dover, 1990:610-611.
- Gorlin R.J., Cohen M.M., Levin L.S. Syndromes of the Head and Neck, NY, Oxford: Oxford University Press, 1990:429-441.
- Jones K.L. Smith’s recognizable Patterns of Human Malformations, Philadelphia, London, Torornto: W.B.Saunders Company, 1988:430-431.
- Warkany J. Congenital malformations. Notes and Comments. Chicago: Year book medical publishers, 1971:1186-1189.
- С.И. Козлова, Н.С. Демикова, Е. Семанова, О.Е. Блинникова. Наследственные синдромы и медико-генетическое консультирование.- М.: Практика, 1996.- 416 с.
Переглянуто редакційною колегією I.B.I.S.: 17/11/2000
Дивіться також:
Програма раннього втручання для дітей із затримкою розвитку “Маленькі сходинки”
Мойра Пітерсі та Робін Трелоар
Університет Маккуорі, Сідней, Австралія

Переклад з англійської і видання здійснено за сприяння Міжнародного благодійного фонду “ОМНІ-Мережа для дітей” та Українського фонду допомоги дітям-сиротам з особливими потребами, Вірджинія, США.
Англійське видання: Pieterse M, Treloar R. Small Steps: An Early Intervention Program for Children With Developmental Delays. Macquarie University, Sydney 1989.
Це програма раннього втручання для стимуляції дітей із затримкою розвитку від народження до 4-річного віку. “Маленькі сходинки” насамперед призначені для батьків дітей з розумовим відставанням. Вони містять практичні поради щодо навчання дітей у домашніх умовах. Основним постулатом цієї програми є те, що батьки – це найкращі вчителі для своєї дитини. Ця програма також може використовуватися педагогами, лікарями і студентами.
- ВІДЕОПРЕЗЕНТАЦІЇ:
- ЗМІСТ:
- Книга 1: Вступ до програми.
Ця вступна книга знайомить вас з історією написання “Маленьких сходинок” і пропонує способи користування програмою.
Завантажити книгу в форматі pdf (Acrobat Reader, 695 кб)
- Книга 2: Програма вашої дитини.
Книга пояснює, як правильно обирати індивідуальні цілі для конкретної дитини, і як допомогти їй досягти цих цілей в оточенні сім’ї.
Завантажити книгу в форматі pdf (Acrobat Reader, 3,51 мб)
- Книга 3: Навички спілкування.
У книзі ми зупинимось на формуванні мовних навичок як на довербальному, так і вербальному рівнях розвитку.
Завантажити книгу в форматі pdf (Acrobat Reader, 75,8 мб)
- Книга 4: Навички загальної моторики.
У книзі описуються навички, які потребують використання великих м’язів тіла. За допомогою цих м’язів дитина може сидіти, повзати, ходити, лазити, ловити м’яч і т.д. Книга підготовлена фізіотерапевтом, який має досвід роботи з маленькими дітьми з вадами розвитку.
Завантажити книгу в форматі pdf (Acrobat Reader, 4,40 мб)
- Книга 5: Навички тонкої моторики.
Навички тонкої моторики використовують дрібні м’язи рук та очей. Ці навички дуже різноманітні і коливаються від здатності дитини схопити палець мами до таких складних умінь, як малювання та різання. У цю книгу також увійшли навички вирішення проблем і розвиток таких понять, як колір, форма і розмір.
Завантажити книгу в форматі pdf (Acrobat Reader, 1,42 мб)
- Книга 6: Сприйняття мовлення.
Навички сприйняття мовлення означають розуміння мови інших. Вони означають здатність дитини уважно слухати, що кажуть інші, слідувати вказівкам, і, що найбільш важливо, самій використовувати мову.
Завантажити книгу в форматі pdf (Acrobat Reader, 872 кб)
- Книга 7: Самообслуговування та соціальні навички.
У цій книзі ми розглянемо навички, які допомагають дитині встановити зв’язок з іншими, гратися і самостійно їсти, користуватися туалетом, одягатися і приводити себе в порядок.
Завантажити книгу в форматі pdf (Acrobat Reader, 1,08 мб)
- Книга 8: Перелік вмінь розвитку дитини.
У книзі об’єднані в одне ціле всі окремі частини “Маленьких сходинок” і представлений перелік методів, що були використані для оцінки дитини. Крім того, вона допоможе вам правильно намітити цілі.
Аміоплазія – показники ваги
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”
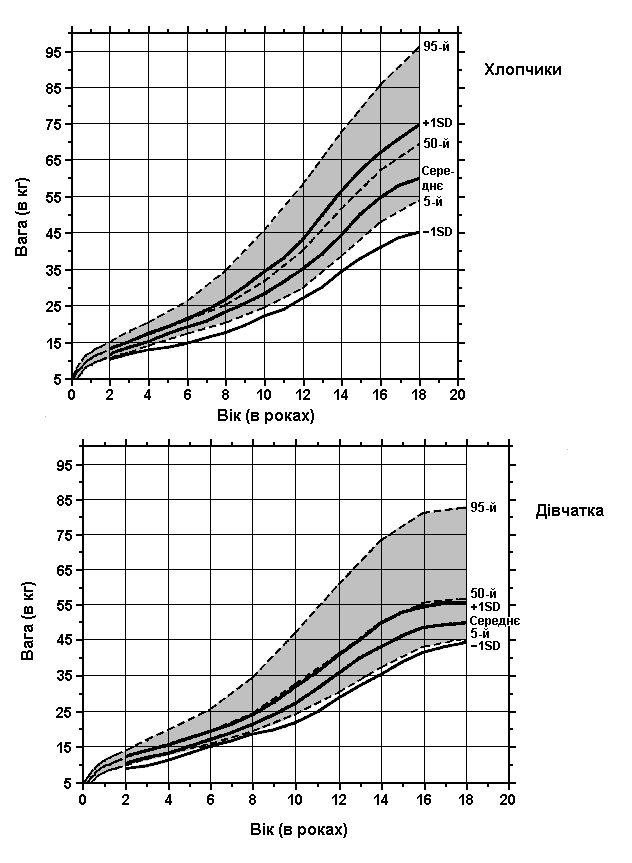 Показники ваги хлопчиків та дівчаток (всього 98 пацієнтів) з аміоплазією порівнюються з результатами обстеження здорових осіб (зафарбована ділянка). Hall JG: Prog Clin Biol Res 200:155, 1985.
Показники ваги хлопчиків та дівчаток (всього 98 пацієнтів) з аміоплазією порівнюються з результатами обстеження здорових осіб (зафарбована ділянка). Hall JG: Prog Clin Biol Res 200:155, 1985.
Завантажити діаграму в форматі pdf (Acrobat Reader, 160 кб)
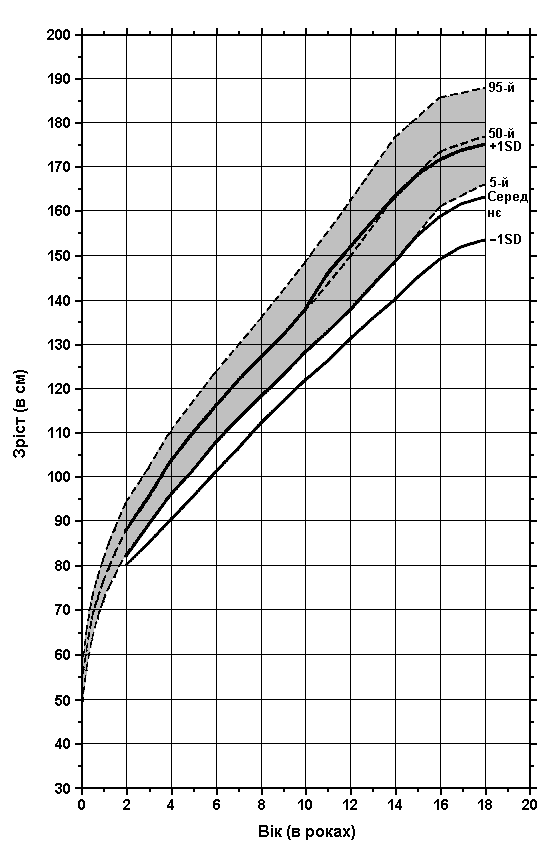
Аміоплазія – зріст у дівчат
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”
Дані обстеження 98 пацієнтів з аміоплазією порівнюються з результатами обстеження здорових осіб (зафарбована ділянка). Hall JG: Prog Clin Biol Res 200:155, 1985.
Завантажити діаграму в форматі pdf (Acrobat Reader, 152 кб)
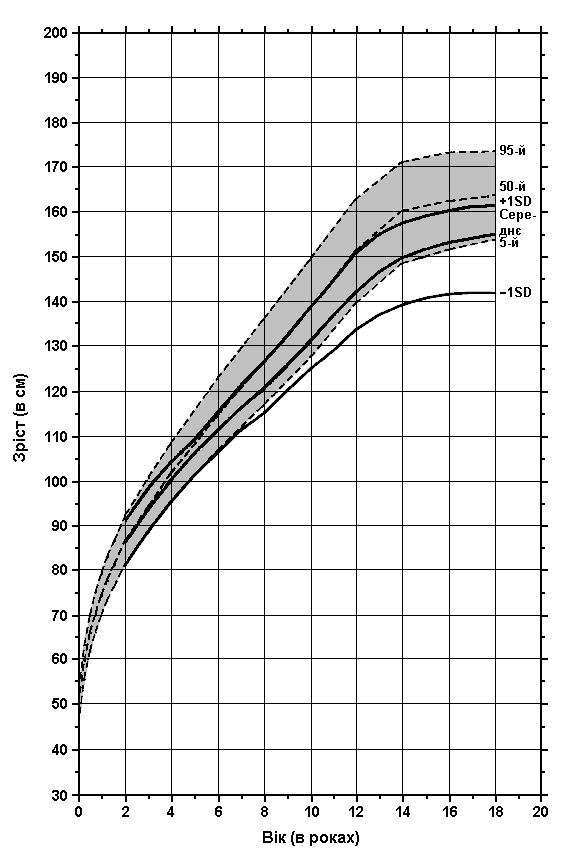
Аміоплазія – зріст у хлопчиків
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”.
Дані обстеження 98 пацієнтів з аміоплазією порівнюються з результатами обстеження здорових осіб (зафарбована ділянка). Hall JG: Prog Clin Biol Res 200:155, 1985.
Завантажити діаграму в форматі pdf (Acrobat Reader, 153 кб)
Синдром Елерса-Данлоса
(Ehlers Danlos Syndrome)
Наталія Зимак-Закутня
Завідуюча Хмельницькою
медико-генетичною консультацією
Синдром Елерса–Данлоса (ЕДС) – гетерогенна група спадкових захворювань сполучної тканини, які характеризуються підвищеною рухливістю суглобів, еластичністю шкіри та крихкістю тканин.
Перший опис синдрому Елерса-Данлоса датується ще в І ст. до н.е. Ґрунтовний клінічний опис належить російському терапевту Черногубову (1892 рік). Але синдром назвали на честь датського дерматолога Едварда Елерса, який вивчав цей синдром у 1901 році та Генрі-Олександра Данлоса, французького лікаря, що проводив експертизу аномалій шкіри у 1908. Саме вони поєднали відомі ознаки цього стану та точно визначили фенотип цієї групи аномалій.
Класифікація:
Перші спроби класифікації ЕДС були здійснені наприкінці 1960-х, у 1968 році під час конференції у Берліні була запропонована найбільш поширена класифікація, що включає принаймні 10 окремих фенотипів ЕДС:
- ЕДС тип І (ОМІМ #130000).
- ЕДС тип ІI (ОМІМ #130010).
- ЕДС тип ІII (ОМІМ #130020).
- ЕДС тип ІV (ОМІМ #130050).
- ЕДС тип V (ОМІМ #305200).
- ЕДС тип VI (ОМІМ #225400).
- ЕДС тип VII (ОМІМ #130060 – типи VІІА і VІІВ, ОМІМ #225410 – тип VІІС).
- ЕДС тип VIII (ОМІМ #130080).
- ЕДС тип IX (ОМІМ #304150).
- ЕДС тип X (ОМІМ #225310).
Нещодавній розвиток у тлумаченні біохімічної та молекулярної основи ЕДС, разом із збільшенням клінічного досвіду, дозволяє удосконалити існуючу класифікацію, роблячи її більш практичною.
У 1998 році Бейтон запропонував спрощену класифікацію, розроблену для полегшення встановлення точного діагнозу синдрому Елерса-Данлоса і чіткішої диференціації аномалій, що перекриваються в існуючій класифікації ЕДС. Нова класифікація ЕДС виділяє 6 основних типів:
- Класичний тип (у минулій класифікації ЕДС – І та ІІ типи).
- Гіпермобільний тип (у минулій класифікації ЕДС – ІІІ тип).
- Судинний тип (у минулій класифікації ЕДС – IV тип).
- Кіфосколіозний тип (у минулій класифікації ЕДС – VI тип).
- Артрохалазійний тип (у минулій класифікації ЕДС – VIIB).
- Дерматоспараксичний тип (у минулій класифікації ЕДС – VIIС).
Типи, які не включені до нової класифікації, наступні:
- V та Х типи ЕДС (X-зчеплені) були описані лише в одній родині.
- VIII тип ЕДС схожий на класичний тип, але він, на додачу, проявляється із періодонтальною крихкістю. Це рідкісний тип ЕДС. Існування цього синдрому як окремого об‘єкта не є певним.
- IX тип ЕДС раніше було по-новому визначено як “синдром потиличного рога”.
- Х–зчеплене рецесивне захворювання алельне синдрому Менкеса (OMIM #309400). Х тип ЕДС було описано лише в одній сім‘ї.
- ЕДС ХІ типу, названий “синдромом сімейної суперрухливості суглобів”, раніше було вилучено із класифікації ЕДС. Зв‘язок його із ЕДС ще не уточнено.
Діагностичні критерії:
Для кожного типу нової класифікації визначено перелік головних та другорядних діагностичних критеріїв. Головний критерій має високу діагностичну специфіку, оскільки він є рідкісним при інших станах та у загальної маси населення. Наявність одного чи більше головних критеріїв свідчить про необхідність проведення клінічної діагностики, або, при можливості, високоінформативного та гарантованого лабораторного підтвердження. Другорядний критерій є ознакою меншої діагностичної специфічності. Наявність одного чи більше другорядних діагностичних критеріїв допомагає при визначенні конкретного типу ЕДС. Між тим, при відсутності головного критерію вони не є достатніми для встановлення діагнозу. Наявність другорядних критеріїв може стати припущенням для встановлення станів пов‘язаних із ЕДС, природа яких буде уточнена після визначення молекулярної основи.
1. Гіпереластичність шкіри повинна перевірятися на нейтральному місці, тобто там, де шкіра не підпадає механічній дії чи рубцюванню, наприклад на нижній поверхні передпліччя. Вона вимірюється шляхом розтягнення шкіри до відчуття опору. У дітей еластичність шкіри виміряти важко у зв‘язку із переважаючою кількістю підшкірного жиру.
2. Гіперрухливість суглобів пропонується оцінювати за допомогою шкали Бейтона. Гіперрухливість суглобів залежить від віку, статі, сімейного анамнезу та етнічного походження. Оцінка 5 із 9 чи більше свідчить про гіперрухливість. Загальна оцінка отримується наступним чином:
- пасивний вигин мізинців більше, ніж на 90° – один бал за кожну руку;
- пасивне згинання великих пальців рук до згинаючого м‘яза передпліччя – один бал за кожну руку;
- гіперекстензія ліктів більше ніж на 10° – один бал за кожен лікоть;
- гіперекстензія колін більше ніж на 10° – один бал за кожне коліно;
- згинання тулуба вперед із повністю випрямленими колінами так, що долоні повністю лежать на підлозі – один бал
3. Легка поява синців свідчить про спонтанні крововиливи, які часто з‘являються в одних і тих самих місцях, що викликає характерну коричневу зміну кольору шкіри. Легка поява синців як симптом може проявлятися у ранньому дитинстві. Жорстоке поводження із дітьми повинне розглядатися як диференційний діагноз. Існує тенденція до тривалої кровотечі замість нормального зсідання крові.
4. Крихкість тканин проявляється легкою появою синців та наявністю дистрофічних шрамів. Шрами з‘являються переважно на точках тиску (наприклад, колінах, ліктях, чолі, підборідді) та мають тонкий, схожий на папір вигляд. Часто шрами стають широкими та безбарвними; рани загоюються важко.
5. Пролапс мітрального клапану (ПМК) та проксимальна дилятація аорти повинні діагностуватися шляхом ехокардіографії, комп‘ютерної томографії та МРТ. Пролапс мітрального клапану є звичним проявом, а дилятація аорти (коли максимальний діаметр синусів перевищує верхню межу норми для певного віку та тілобудови) – рідкісним; у незначної кількості пацієнтів із ЕДС вона може бути прогресуючою. Для встановлення діагнозу ПМК повинні застосовуватися чіткі критерії. У тих пацієнтів, які мають дилятацію аорти, при диференційному діагнозі необхідно звернути увагу на розширення кільця аорти.
3. Хронічні болі у суглобах та кінцівках є частим симптомом і потребують Re-графії. Також проблеми викликає чітке визначення анатомічної локалізації болю.
Опис типів синдрому:
Хоча і чітко окреслені, кіфосколіозний, артрохалазійний та дерматоспараксичний типи трапляються набагато рідше, ніж класичний, гіпермобільний та артеріальний типи.
I. Класичний тип.
1. Успадкування:
- Аутосомно–домінантний тип.
2. Головні діагностичні критерії:
- Гіперрозтяжність шкіри.
- Поширені атрофічні шрами (ознака крихкості тканин).
- Гіперрухливість суглобів.
3. Другорядні діагностичні критерії:
- Гладка, бархатиста шкіра.
- Несправжні молюскоїдні пухлини.
- Підшкірні сфероїди.
- Ускладнення внаслідок гіперрухливості суглобів (наприклад, розтягнення, зміщення/підвивих, пласка ступня).
- М’язова гіпотонія, затримка моторного розвитку.
- Легке виникнення синців.
- Прояви шкірної розтяжності та крихкості (наприклад, хіатальна грижа, пролапс у дитинстві, цервікальна недостатність).
- Хірургічне ускладнення (післяопераційні грижі).
- Позитивний сімейний анамнез.
4. Причини та лабораторна діагностика:
- При класичному типі ЕДС у певній кількості сімей, але не у всіх, було визначено порушення електрофоретичної рухливості ланцюгів колагену proα1(V) або proα2(V) V типу. У зв‘язку із тим, що високочутливий метод скринінгу ще не розроблений, відсутність виявлених відхилень при біохімічному та молекулярному аналізі не виключає наявність дефекту колагену V типу. В інформативних сім’ях для пренатального та постнатального діагнозу може застосовуватися дослідження генетичного зчеплення. Аналіз мутацій у окремих осіб проводиться на дослідницькій основі.
5. Спеціaльні зауваження:
- Шкірні прояви можуть мати різний ступінь важкості; було описано родини із м‘яким, помірним та важким ступенем прояву.
- Несправжні молюскоїдні пухлини – це ураження шкіри, пов‘язані із шрамами. Вони часто трапляються у місцях, які зазнають тиску, наприклад, ліктях.
- Сфероїди – це дрібні підшкірні тверді утворення, часто рухливі та відчутні на дотик на передпліччях та гомілках. Сфероїди можуть твердішати та бути визначеними на рентгені.
- Постійні підвивихи суглобів у плечових, колінних та скронево-нижньощелепних суглобах.
- Диспареунія та статеві розлади іноді зустрічаються при класичній та інших формах ЕДС.
- Частою скаргою є втомлюваність.
II. Гіпермобільний тип.
1. Успадкування:
- Аутосомно–домінантний тип.
2. Головні діагностичні критерії:
- Ураження шкіри (гіперрозтяжність та/або гладка, бархатиста шкіра).
- Генералізована гіпермобільність суглобів.
3. Другорядні діагностичні критерії:
- Постійні підвивихи суглобів.
- Хронічні болі у суглобах/кінцівках.
- Позитивний сімейний анамнез.
4. Особливі зауваження:
- Ступінь розтяжності шкіри різний. Наявність атрофічних шрамів у осіб із гіперрухливістю суглобів вказує на можливий діагноз класичного типу.
- Гіперрухливість суглобів є домінуючим клінічним проявом. Окремі суглоби, такі як плечові, колінні та скронево–нижньощелепні суглоби, часто зміщуються.
- У практиці ревматології, значна кількість пацієнтів також має генералізовану гіперрухливість суглобів. Важливо відрізнити цих осіб від хворих на ЕДС гіпермобільного типу. Спірним питанням наразі є причинні взаємозв‘язки, якщо вони взагалі існують, між фенотипами таких осіб та хворих на гіпермобільний тип ЕДС.
- М‘язово-скелетні болі виявляються рано, є хронічними та, можливо, ослаблюючими (викликають слабкість). Анатомічне розповсюдження широке, іноді можуть виявлятися слабкі місця. Слабке місце визначається, як болюче місце при пальпації великим, 2 чи 3 пальцями при тиску в 4 кг чи менше.
III. Судинний тип.
Судинний тип ЕДС спричинений структурними дефектами у ланцюгу колагену proa1(III) ІІІ типу, кодований COL3A1.
1. Успадкування:
- Аутосомно–домінантний тип.
2. Головні діагностичні критерії:
- Тонка, напівпрозора шкіра.
- Артеріальна/кишкова/маткова крихкість чи розриви.
- Поширені кровотечі.
- Характерний зовнішній вигляд.
3. Другорядні діагностичні критерії:
- Акрогерія.
- Гіперрухливість малих суглобів.
- Розриви сухожиль та м‘язів.
- Квіно-варусна клишоногість.
- Ранній прояв варикозного розширення вен.
- Артеріо-венозні, каротидно-кавернозні фістули.
- Пневмоторакс/пневмогемоторакс.
- Атрофія ясен гінгівального краю.
- Позитивний сімейний анамнез, раптова смерть у близького родича (родичів).
Наявність двох або більше головних критеріїв вказує зі значною вірогідністю на діагноз, при цьому настійливо рекомендовані лабораторні дослідження.
4. Причини та лабораторні дослідження:
- Методика проведення лабораторного дослідження включає: 1) виявлення структурно зміненого колагену ІІІ типу, який виробляється фібробластами, що спричиняє порушення секреції, посттрансляційну модифікацію, температурну нестійкість, та/чи чутливість до протеаз, 2) виявлення мутацій у гені COL3A1. Визначення рівня проколагену ІІІ типу у сироватці крові у клінічній практиці не є доступним.
5. Окремі зауваження:
- У деяких уражених осіб присутні характерні риси зовнішності.
Відзначається збільшення підшкірної жирової тканини, особливо на обличчі та кінцівках. - Гіпермобільність суглобів, як правило, обмежена.
- Спонтанний розрив артерій найчастіше трапляється у 3 та 4 десятиліттях життя, але може відбутися і раніше. Артерії середнього розміру уражаються найчастіше. Артеріальні розриви є найчастішою причиною раптової смерті.
- Гострі болі у животі та в боку (розлиті чи локалізовані) звичайно виявляються внаслідок артеріальних чи кишкових розривів, тому на предмет цього необхідно негайно проводити огляд. Рекомендуються неінвазивні діагностичні процедури.
- Підшкірний венозний малюнок особливо помітний на грудній клітині та животі.
- За наявності серйозних синців як початкового ускладнення, необхідно виключити можливу причину у жорстокому поводженні з дітьми чи гематологічних розладах. У цьому контексті при хронічних синцях та утвореннях шрамів, необхідно провести диференційну діагностику із класичним типом ЕДС.
- Діагностика цього стану у дітей викликає скадність при відсутності сімейного анамнезу.
- Як ускладнення вагітності може виникнути інтранатальний розрив матки та пре- і постнатальна кровотеча. Під час пологів можуть трапитися вагінальні та перинеальні розриви.
- Під час та після хірургічного втручання часто трапляються серйозні ускладнення (наприклад розходження рани).
IV. Кіфосколіозний тип.
Причиною є недостатність лізилгідроксилази (PLOD), колагензмінюючого ензиму внаслідок гомозиготності чи складної гетерозиготності у мутантному аллелі (аллелях) PLOD.
1. Успадкування:
- Аутосомно–рецесивний тип.
2. Головні діагностичні критерії:
- Генералізована слабкість суглобів.
- Важка м‘язова гіпотонія при народженні.
- Вроджений прогресуючий сколіоз.
- Крихкість склер та розриви очного яблука.
3. Другорядні діагностичні критерії:
- Крихкість тканин, включно із атрофічними шрамами.
- Виникнення синців.
- Артеріальні розриви.
- Марфаноїдний зовнішній вигляд.
- Мікрокорнеа.
- Значний остеопороз при рентгенівському дослідженні.
- Сімейний анамнез, тобто наявність уражених сибсів.
Наявність трьох головних діагностичних критеріїв у дитини є основою для діагностики і проведення відповідних лабораторних досліджень.
4. Причини та лабораторні дослідження:
- Доступним, високочутливим та точним методом є вимірювання загального рівня гідроксилізил-пирідиноліну та лізил-пирідиноліну у сечі після гідролізу методом рідинної хроматографії високого тиску (РХВТ). Визначення дермального гідроксилізину також не викликає труднощів, хоча, визначення активності лізилгідроксилази у фібробластах та/чи аналіз мутацій гена PLOD проводиться поки що експериментально.
5. Окремі зауваження:
- М‘язова гіпотонія може бути яскраво вираженою, це веде до затримки загального моторного розвитку. На цей момент необхідно звернути увагу при початковому диференційному діагнозі у дітей із гнучкістю.
- Фенотип найчастіше складний, нерідко відбувається втрата можливості ходити у 2 та 3 декадах.
- Склеральна крихкість може призвести до розривів очного яблука навіть після дрібної травми. Цей стан необхідно диференційювати із синдромом крихкої рогівки. Наразі відомо, що серйозні ускладнення із очима трапляються набагато рідше ніж вважалося до цього, звідси і відповідні зміни у описі даного типу.
- Складна неонатальна форма синдрому Марфана повинна розглядатися як диференційний діагноз.
- Є дані про менш важку форму цього стану, з нормальною активністю лізил гідроксилази та нормальним вмістом гідроксилізину у шкірі (OMIM #229200); ця форма є ще рідкіснішою.
V.Артрохалазійний тип.
Викликається мутацією, яка веде до порушення продукування амінотермінального кінця ланцюгів proa1(I) (тип A) чи proa2(I) (тип Б) колагену типу І пов‘язаного із переміщенням екзону 6 у інший ген.
1. Успадкування:
- Аутосомно–домінантний тип.
2. Головні діагностичні критерії:
- Складна генералізована гіпермобільність суглобів із постійними підвивихами.
- Вроджений двосторонній вивих стегна.
3. Другорядні діагностичні критерії:
- Гіперрозтяжність шкіри.
- Крихкість тканин, включно із атрофічними шрамами.
- Легке виникнення синців.
- М‘язова гіпотонія.
- Кіфосколіоз.
- М‘яка форма остеопорозу при рентгенівському дослідженні.
4. Причини та лабораторні дослідження:
- Біохімічний дефект визначається шляхом електрофоретичної детекції ланцюгів pNα1(I) чи pNα2(I) дермального колагену.
5. Окремі зауваження:
- Вроджений вивих стегна спостерігався у всіх пацієнтів, у яких біохімічні аналізи були позитивними.
- Низький зріст не є проявом, якщо тільки він не є ускладненням важкої форми кіфосколіозу та/чи вивиху стегна.
- У якості диференційного діагнозу слід розглядати синдром Ларсена.
VI. Дерматоспараксичний тип.
Спричиняється недостатністю N-термінальної пептидази проколагену І, викликаною гомозиготністю чи компаундною гетерозиготністю мутантних аллелів (на відміну від артрохалазійного типу, який є результатом мутацій субстратних сайтів із залученням ланцюгів проколагену І типу.
1. Успадкування:
- Аутосомно–рецесивний тип.
2. Головні діагностичні критерії:
- Виражена крихкість шкіри.
- Обвисаюча, зайва шкіра.
3. Другорядні діагностичні критерії:
- М‘яка, рихла текстура шкіри.
- Легка поява синців.
- Передчасні розриви ембріональних мембран.
- Великі кили (пуповинні, пахові).
4. Причини та лабораторні дослідження:
- Біохімічно підтверджується електрофоретичною детекцією ланцюгів pNα1(I) чи pNα2(I) колагену типу І, екстрагованих із шкіри в присутності протеазних інгібіторів, чи отриманих із фібробластів. Визначення активності N-протеїнази проводиться лише на експериментальній основі.
5. Особливі зауваження:
- Суттєвою є крихкість шкіри та виникнення синців. Заживлення ран не забруднене без утворення атрофічних шрамів.
- Надлишок шкіри на обличчі призводить до зовнішнього вигляду що нагадує cutis laxa; хоча, виникнення синців та крихкість шкіри не виявляються при cutis laxa.
- Назва взята із схожого фенотипу та біохімічного дефекту, який раніше розпізнавали у великої рогатої худоби, овець та інших тварин.
- Кількість описаних випадків мала і фенотиповий спектр може бути розширений.
|
|
||||
|
|
||||
|
|
||||
|
|
||||
|
|
||||
|
|
Література:
- Козлова С.И., Демикова Н.С., Семанова Е., Блинникова О.Е. Наследственные синдромы и медико-генетическое консультирование.- М.: Практика, 1996.- С.41.
- Beighton P, Grahame R, Bird H. Hypermobility of Joints. 2-nd edition. Berlin: Springer. 1989:1–182.
- Beighton P, De Paepe A, Danks D, Finidori G, Gedde-Dahl T, Goodman R, Hall JG, Hollister DW, Horton W, McKusick VA, Opitz JM, Pope FM, Pyeritz RE, Rimoin DL, Sillence D, Spranger JW, Thompson E, Tsipouras P, Viljoen D, Winship I, Young I. International nosology of heritable disorders of connective tissue, Berlin, 1986. Am J Med Genet. 1988;29:581–594.
- Beighton P, De Paepe A, Steinmann B, et al. Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers- Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK). Am J Med Genet. 1998 Apr 28;77(1):31-7.
- Golfier F, Peyrol S, Attia-Sobol J, et al. Hypermobility type of Ehlers-Danlos syndrome: influence of pregnancies. Clin Genet. 2001 Sep;60(3):240-1.
- Gorlin R.J., Cohen M.M., Levin L.S. Syndromes of the Head and Neck, NY, Oxford: Oxford University Press. 1990:429-441.
- Jones KL. Smith’s Recognizable Patterns of Human Malformation. W.B. Saunders Company, Philadelphia. 1997:430-431.
- Leier CV, Call TD, Fulkerson PK, Wooley CF. The spectrum of cardiac defects in the Ehlers-Danlos syndrome, types I and III. Ann Intern Med. 1980;92:171–178.
- Maltz SB, Fantus RJ, Mellett MM, Kirby JP. Surgical complications of Ehlers-Danlos syndrome type IV: case report and review of the literature. J Trauma. 2001 Aug;51(2):387-90.
- Mao JR, Bristow J. The Ehlers-Danlos syndrome: on beyond collagens. J Clin Invest. 2001 May;107(9):1063-9.
- Pepin MG, Superti-Furga A, Byers PH. Natural history of Ehlers-Danlos syndrome type IV (EDS type IV): Review of 137 cases. Am J Hum Genet. 1992;51:44.
- Roman MJ, Rosen SS, Kramer-Fox R, Devereaux RB. The prognostic significance of the pattern of aortic root dilatation in the Marfan syndrome. J Am Coll Cardiol. 1993;22:1470–1476.
- Sacheti A, Szemere J, Bernstein B, Tafas T, Schechter N, Tsipouras P. Chronic pain is a manifestation of the Ehlers-Danlos syndrome. J Pain Symptom Managem. 1997;14:88–93.
- Schaefer B. Ehlers-Danlos syndrome (www.emedicine.com).
- Sorokin Y, Johnson MP, Rogowski N, Richardson DA, Evans MI. Obstetric and gynecologic dysfunction in the Ehlers-Danlos syndrome. J Reprod Med. 1994;39:281–284.
- Steinmann B, Superti-Furga A, Joller-Jemelka HJ, Cetta G, Byers PH. Ehlers-Danlos syndrome type IV: A subset of patients distinguished by low serum levels of the amino-terminal propeptide of type III procollagen. Am J Med Genet. 1989;34:68–71.
- Steinmann B, Eyre DR, Shao P. Urinary pyridinoline cross-links in Ehlers-Danlos syndrome type VI. Am J Hum Genet. 1995;57:1505–1508.
- Warkany J. Congenital malformations. Notes and Comments. Chicago: Year book medical publishers. 1971:1186-1189.
Переглянуто редакційною колегією I.B.I.S.: 26/05/2005
Дивіться також:
|
|
|
|
|
|





