
Serhiy
Типи плаценти при багатоплідній вагітності
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”
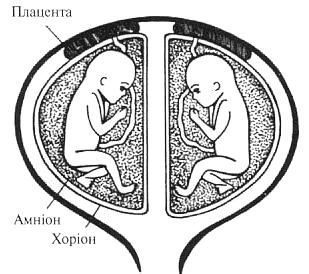
Розділена діхоріонічна діамніотична плацента. Така плацента спостерігається в усіх випадках дізиготної вагітності та у монозиготній вагітності внаслідок розділення бластомерів протягом трьох днів після запліднення. Якщо зиготи-близнюки прикріпляються на різних сторонах стінки матки, то диски плаценти будуть повністю відокремленими.
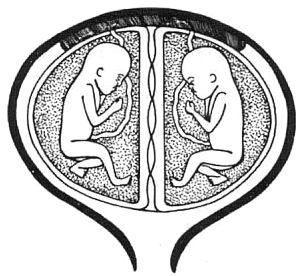
Зрощена діхоріонічна діамніотична плацента. Якщо зиготи-близнюки прикріпляються близько одна до одної на стінці матки, то між дисками плаценти може бути зрощення різного ступеня.
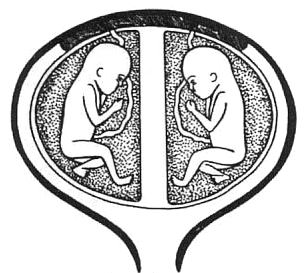
Монохоріонічна діамніотична плацента спостерігається у випадках, коли утворення монозиготних близнюків відбувається між четвертим та восьмим днем після запліднення.
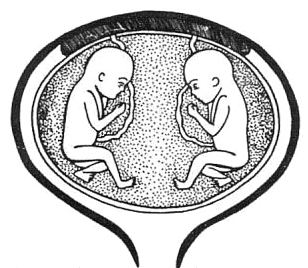
Монохоріонічна моноамніотична плацента спостерігається у випадках, коли утворення близнюків відбувається після восьмого дня запліднення. Плоди-близнюки знаходяться в одній амніотичній порожнині.
Взято з Human Malformations and Related Anomalies by Roger Stevenson and Judith Hall, et al. Copyright, 1993 by Oxford University Press. Використовується з дозволу Oxford University Press.
Пілоростеноз (інформація № 2)
(Pyloric Stenosis)
Георгій Леонідович Лінчевський
Доцент, зав. курсом неонатології,
Донецький державний медичний університет ім. М. Горького
Визначення:
Пілоростеноз – вроджене звуження пілоричного відділу шлунка.
Етіологія та патогенез:
Пілоростеноз зумовлений комплексним впливом спадкових чинників та факторів зовнішнього середовища, які умовно можна згрупувати наступним чином:
Ендогенні фактори:
- зміни генетичних структур (генні мутації і хромосомні аберації);
- ендокринні захворювання;
- “перезрівання” статевих клітин;
- вік батьків.
Екзогенні фактори:
- Фізичні фактори:
- радіаційні;
- механічні.
- Хімічні фактори:
- лікарські препарати;
- хімічні речовини, які застосовуються в побуті та промисловості;
- гіпоксія;
- неповноцінне харчування.
- Біологічні фактори:
- віруси;
- мікоплазми;
- протозойні інфекції;
- ізоімунізація.
Мультифакторні.
У багатьох випадках множинних вад, що включають пілоростеноз, виявлені хромосомні (трисомії 18 і 21, часткові трисомії 1d, 5d, 13d; часткові моносомії 15d і 21d) і моногенні синдроми, як з аутосомно-домінантним (Альпорта, Пфайффера), так і з аутосомно-рецесивним успадкуванням (Сміта-Лемлі-Опітца).
Ізольовані форми вродженого пілоростенозу частіше мають мультифакторіальне походження (Schafer K., Kramer M., 1987 р.).
Патогенез:
Патогенез даної патології відмінний для різних її видів. Морфологи виділяють 2 типи пілоростенозу:
- Тип Mayer-Landerer: вада, що виникає внаслідок порушення зворотного розвитку центральної частини ентодермального епітелію, який спостерігається у зародка в 5-6 тижнів і вростання в нього з периферії навколишньої мезенхіми на 7-8 тижні, що утворює строму слизової оболонки з ворсинками. У результаті це призводить до звуження на даній ділянці травної трубки.
- Тип Гіршпрунга: стенозуюча гіпертрофія пілоричного відділу шлунку. Причина і механізм розвитку захворювання до кінця не з’ясовані. Більшість дослідників сходяться на думці, що в основі лежить гіпертрофія м’язового шару пілоричної частини шлунку, обумовлена порушенням її інервації. Певний інтерес викликає теорія Томсона, у якій основна роль відводиться частим спазмам пілоричного відділу шлунку, що призводять до робочої гіпертрофії її м’язового шару (мал. 1).

Гіпертрофічний пілоростеноз (Т.Е. Іванівська, А.В. Цинзерлінг, 1976).
а – різка гіпертрофія м’язового шару з утворенням пілоричного каналу
в хлопчика 1 місяця 9 днів; б – нормальний пілоричний сфінктер шлунку
в хлопчика 6 місяців. Фарбування гематоксиліном і еозином. х100
Клінічна картина:
Основними симптомами є:
- блювота фонтаном, що виникає з 2-4-тижневого віку майже після кожного годування;
- перистальтика шлунку у вигляді двох округлих випинань, що нагадують пісочний годинник;
- закрепи;
- зменшення кількості сечовиділень;
- швидке зниження маси тіла; через 5-6 тижнів після початку захворювання втрата маси тіла досягає 75-80% маси при народженні;
- відзначається млявість, сонливість, різке виснаження.
Блювота є одним із найважливіших симптомів пілоростенозу. Вона виникає до кінця 2-3 тижня життя, в окремих випадках – з перших днів життя, або пізніше (у 1-1,5 місяця). Блювотні маси майже після кожного годування викидаються з великою силою. Їхня кількість через надмірну секрецію шлункового соку буває значно більшою, ніж кількість рідини, спожитої дитиною. У блювотних масах ніколи не буває жовчі. При тривалому перебігу хвороби слиз, що виявляється в блювотних масах, має запах низькомолекулярних жирних кислот, що свідчить про атонію і дилятацію шлунку. У 93% дітей до початку блювоти відмічається зригування, що не відображається на загальному стані дитини. Цей початковий період захворювання, що триває від 3-4 до 7 днів, у більшості випадків залишається непоміченим. У перші 2 тижні хвороби блювота носить стійкий характер, виникає через 10-15 хвилин після кожного годування. У наступні дні вона трапляється рідше, виникає в більш пізній термін після закінчення годування хворого. Дитина в цей період відчуває біль (страждальницький вираз обличчя, лемент).
Перистальтика шлунку. Перистальтика у вигляді пісочного годинника, що виявляється при годуванні чи поверхневій пальпації живота дитини, є також основним симптомом пілоростенозу, зустрічається у 80% дітей з цим захворюванням, у багатьох ці симптоми бувають стертими. Іноді вони відсутні навіть у тих хворих, у яких добре виражені інші симптоми пілоростенозу.
Втрата ваги. З початком блювоти поступово зменшується вага дитини. У деяких випадках втрата ваги відносно ваги при народженні сягає 30%. Однією з головних причин, крім стенозу воротаря, є ступінь спазму цього відділу. При невираженому спазмі відмічається незначне зниження ваги, при вираженому – значна втрата.
Затримка випорожнень і зменшення кількості сечовипускань. Затримка випорожнень спостерігається у дітей з перших днів появи блювоти. До її виникнення діти мають нормальні щоденні випорожнення. Псевдообстипація починається внаслідок недостатнього надходження їжі в кишковий тракт. У дітей з пілоростенозом колір випорожнень нагадує колір меконію. Причина такого забарвлення випорожнень – мала кількість їжі, що надходить у тонкий кишечник, при наявності нормального надходження жовчі. Зрідка у пізніших термінах захворювання відзначаються диспепсичні чи “голодні” випорожнення. При пілоростенозі зменшення числа сечовипускань і кількості сечі є важливими симптомами. У деяких випадках кількість сечі не перевищує 16 мл на добу замість 300-400 мл, що виділяє дитина у віці 1-1,5 місяці.
Діагностичні підходи:
Лабораторні дослідження. При пізньому встановленні діагнозу внаслідок значного зневоднення організму, викликаного постійною блювотою, відмічається згущення крові, що проявляється у підвищенні кількості гемоглобіну й зниженні ШОЕ.
Гіпохлоремія у дітей з пілоростенозом є одним з важливих лабораторних симптомів, вона може стати причиною виникнення гіпохлоремічної коми.
Рентгенологічне дослідження проводять не раніше ніж через 5-6 годин після останнього годування. Після оглядової рентгеноскопії грудної клітки і черевної порожнини дитина одержує з ріжка барій у вигляді 5% суспензії в грудному молоці не більше 50-60 мл. Після прийому барію рентгеноскопія проводиться у фронтальній та сагітальній проекції. У нормі пілоричний жом відкривається через 5 хвилин. При пілоростенозі цей термін подовжується до 30 хвилин.
При рентгенологічному дослідженні з барієм звертають увагу на 4 основних симптоми:
- розмір шлунку;
- його перистальтику;
- час випорожнення шлунку;
- звуження і подовження пілоричного каналу.
Розмір шлунку. Шлунок у результаті затримки їжі та гіперсекреції найчастіше розтягнутий. Оцінюючи це розширення, враховують ряд моментів:
- розмір шлунку в однієї дитини може змінюватися залежно від того, у якому стані знаходиться шлунок: скорочення чи розслаблення. Тому оцінку розмірів шлунку можна робити тільки спостерігаючи за ним протягом усього періоду рентгенологічного дослідження;
- розширення шлунку, як рентгенологічний симптом при пілоростенозі, спостерігається в основному тільки після 2-3-тижневого терміну захворювання, тобто у відносно пізній термін хвороби. У більш ранній термін захворювання це розширення трапляється зрідка.
Перистальтика шлунку при пілоростенозі різко посилена. Глибока сегментуюча перистальтика по типу “пісочного годинника” є найбільш типовою. Вона добре виражена у дітей в різний термін захворювання.
Випорожнення шлунку. Повне випорожнення шлунку в нормі відбувається в межах 2,5-6 годин, причому починається в перші ж хвилини після прийому барієвої суспензії. При пілоростенозі випорожнення починається тільки через 15-20 хвилин і, залежно від ступеня звуження пілоричного каналу і сили спазму, продовжується 6-10 годин. Залишки барію зберігаються в шлунку до 24 годин.
Звуження і подовження пілоричного каналу. При значному стенозі пілоруса можуть бути виявлені наступні зміни:
- витягнутість антрум у вигляді дзьоба, тонкої стрічки;
- вдавления по краю антрум, пов’язані з гіпертрофією пілоричного відділу;
- подовжений канал воротаря у вигляді тонкої лінії;
- подовжений, не заповнений барієм проміжок між антрум і дванадцятипалою кишкою;
- заповнення барієм тільки дуоденального ковпачка;
- уповільнений початок випорожнення шлунку.
У практичній роботі прийнято вважати, що якщо через 3 години після введення рентген-контрастної речовини вона відсутня у дванадцятипалій кишці, чи якщо через 24 години значна частина її залишається в шлунку, можна з упевненістю ставити діагноз пілоростенозу.
Ультразвукове дослідження. Ехографічна картина пілоростенозу характеризується потовщенням м’язового шару, подовженням пілоричного каналу і порушенням евакуації харчових мас з шлунку. При поперечному скануванні пілоричний відділ має вигляд круглого утвору, що складається з гіпоехогенного вінця і гіперехогенного центру – “пальчик”, “бичаче око”. При поздовжньому скануванні пілоричний відділ візуалізується як циліндричне утворення із потовщеними стінками до 4 і більше міліметрів (див мал. 2).

Мал. 2. Пілоростеноз.
Між голівкою підшлункової залози та воротами печінки
при поздовжньому скануванні візуалізується потовщений
пілорус. Загальний його діаметр до 15 мм, товщина до 7 мм,
довжина 22 мм. При поперечному скануванні візуалізується
округлий утвір з товстим гіпоехогенним вінцем – м’яз
із центром підвищеної ехогенності
Ендоскопічне обстеження проводиться дитячим фібробронхоскопом з діаметром тубуса не більше 5 мм через 5-6 годин після останнього годування. Підготовка до фіброезофагогастродуоденоскопії полягає в пероральній атропінізації на протязі 2-3 днів. Перед обстеженням проводиться контрольне зондування шлунку. Обстеження проводиться після попередньої премедикації під глибокою седацією чи загальним знечуленням. Ендоскопічні ознаки пілоростенозу:
- різке звуження пілоричного відділу шлунка з ригідними зморшками у вигляді “розетки”, або “лійки”, або асиметричного звуження;
- можливе пролабування гіпертрофованих складок слизової в просвіт антрума;
- відсутність перистальтичного скорочення воротаря;
- отвір воротаря діаметром 0,1-0,3 см, непрохідний для тубуса ендоскопу;
- при інсуфляції повітря чи механічному подразненні воротаря, останній не розкривається, форма зморшок не змінюється.
Диференціальний діагноз:
Практично найбільш важко віддиференціювати вроджений пілоростеноз від пілороспазму. Основними диференційно-діагностичними симптомами є:
- втрата ваги;
- наявність чи відсутність перистальтики шлунку;
- терміни початку випорожнювання шлунка при рентгенологічному обстеженні з барієм.
Вага дитини, що хворіє на пілоростеноз, у більшості випадків, різко знижується. При пілороспазмі значного зменшення ваги не відзначається. Виражена перистальтика шлунку у вигляді пісочного годинника виявляється лише при важких формах пілороспазму. Під час рентгенологічного обстеження ця перистальтика носить несегментуючий, а хвилеподібний характер. Випорожнення звичайно починається відразу після прийому їжі, і барій затримується в шлунку від 3 до 10 годин.
В окремих випадках, при стертій клінічній симптоматиці, особливо при легких формах пілоростенозу, з метою диференціальної діагностики доцільно провести курс лікування спазмолітичними засобами (атропін 1:1000 по 3 краплі 3 рази в день, піпольфен, аміназин протягом 5-7 днів і т.п. ). Відсутність успіху лікування свідчить про більшу вірогідність пілоростенозу, ніж пілороспазму.
Синдром Дебре-Фібігера. Перші симптоми цього захворювання з’являються в ті ж терміни, що і при пілоростенозі – на 3-4-му тижні життя. Головними клінічними симптомами його є: а) блювота, часто фонтаном, іноді непостійна, в окремих випадках з домішкою жовчі; б) втрата маси тіла; в) адинамія; г) ексикоз; д) схильність до проносів. Під час рентгенологічного обстеження затримка барію в шлунку не спостерігається. При біохімічному аналізі крові виявляється гіперкаліємія (при пілоростенозі – гіпокаліємія). З клінічних симптомів необхідно звертати увагу на мінливість блювоти, іноді наявність у блювотних масах домішок жовчі та, що особливо важливо, відмову дітей від прийому рідини (на відміну від дітей з пілоростенозом, які жадібно п’ють).
Пілоростеноз також диференціюють з незавершеним поворотом кишечника і мегадуоденумом. Перші симптоми незавершеного повороту кишечника виникають здебільшого на 5-7 добу життя, а в окремих дітей – і в більш пізній термін. На відміну від пілоростенозу в блювотних масах у меншій чи більшій кількості завжди виявляються домішки жовчі. Блювота має непостійний характер.
При обстеженні шлунково-кишкового тракту з барієм незавершений поворот кишечника має характерну рентгенологічну картину. Випорожнення шлунку починається з перших хвилин обстеження, рух контрастної речовини в тонкий кишечник відбувається дуже повільно. Через 15-30 хвилин після прийому барієвої суспензії видно розширену частину вертикальної чи горизонтальної ділянок дванадцятипалої кишки і барій у тонкому кишечнику. При введенні повітря в товстий кишечник часто спостерігається аномалія розташування – сліпа кишка розташовується не у правій здухвинній області, а під печінкою, у епігастральній ділянці чи у ділянці пупка.
На відміну від пілоростенозу для вади розвитку дванадцятипалої кишки характерна блювота з домішкою жовчі. Блювота виникає у першу добу життя дитини при повній непрохідності в області дванадцятипалої кишки (атрезія, перетинчаста форма), при стенозі дванадцятипалої кишки – на 3-4 добу (кільцеподібна підшлункова залоза, ембріональні тяжі, мегадуоденум, викликана недостатньою іннервацією в стінці дуоденум). При оглядовому рентгенологічному дослідженні черевної порожнини виявляються два великих рівні рідини: один у шлунку, другий у дванадцятипалій кишці.
Зовнішній вигляд дитини, хворої на пілоростеноз показаний на мал. 3.
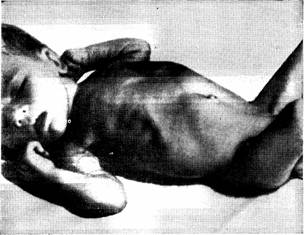
Перешкода в пілоричній ділянці обумовлює глибоку
перистальтику шлунка – симптом пісочного годинника
Лікування:
Оперативне – пілороміотомія по Фреде-Рамштедту, з обов’язковою доопераційною підготовкою від 12 год. до 2 діб з адекватною інфузійною терапією під контролем лабораторних досліджень.
У післяопераційному періоді тактика годування наступна: через 2 години по 7-10 мл 5% розчину глюкози per os, ще через 1 годину 10 мл зцідженого грудного молока, а далі через кожні 2 години по 10 мл грудного молока з подальшим щоденним збільшенням на 100 мл грудного молока на добу.
Через 7 діб прикладати до грудей на 7 годувань.
Паралельно продовжується інфузійна терапія з урахуванням об’єму годування та добової потреби рідини.
Номер з каталогу МІМ:
179010 Pyloric Stenosis, Infantile.
Література:
- Братанов Б. Клиническая педиатрия.- София, 1989.
- Кишковский А. Н. Дифференциальная рентгенодиагностика в гастроэнтерологии.- М.: Медицина, 1984.
- Лазюк Г.И. Тератология человека.- М.: Медицина, 1991.
- Левин Ю. Р. Гастрография у детей раннего возраста.- 1975.
- Ленюшкин А.И. Аномалии развития кишечника.- 1984.
- Михайлов А. Н. Рентгеносемиотика и диагностика болезней человека.- Минск: Вышэйшая школа, 1989.
- Михайлов А. Н. Руководство по медицинской визуализации.- Минск: Вышэйшая школа, 1996.
- Резник Б.Я., Минков И.П. Врожденные пороки развития у детей.- Одесса, 1994.
- Рентгенодиагностика в педиатрии /Под ред. проф. В.Ф. Баклановой, проф. М.А. Филиппкина.- т. I.- М.: Медицина, 1988.
- Ровенская Н.М., Цыбровская Г.Е., Лепихов П.А. Атлас врожденных пороков и дизонтогенетических опухолей внутренних органов у детей.- Донецк: ООО „Лебедь”, 1999.
- Тагер И. Л., Филипкин М. А. Рентгенодиагностика заболеваний органов пищеварения у детей.- М.: Медицина, 1974.
- Шабалов Н.П., Цвелева Ю.В. Основы перинатологии.- М.: МЕДпресс-информ, 2002.
- Шабалов С.Я. Долецкий, В.В. Гаврюшов, В.Г. Акопян. Хирургия новорожденных.- М.: Медицина, 1976.
Переглянуто редакційною колегією I.B.I.S.: 22/01/2004
Дивіться також:
Рентгенографічні ознаки зрілості скелету (частина 4)
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”
Вік скелету (чоловічий) – 156 місяців
Вік скелету (жіночий) – 128 місяців
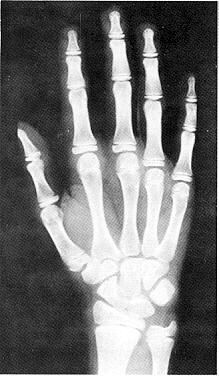
Вік скелету (чоловічий) – 180 місяців
Вік скелету (жіночий) – 148 місяців
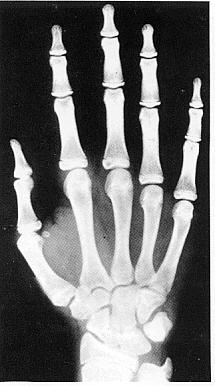
Вік скелету (чоловічий) – 192 місяці
Вік скелету (жіночий) – 161 місяць
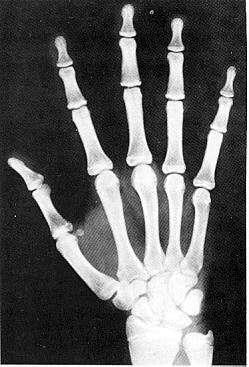
Вік скелету (чоловічий) – 216 місяців
Вік скелету (жіночий) – 192 місяці
Пілоростеноз (інформація № 1)
(Pyloric Stenosis)
М.І. Гогоша
Головний педіатр
Волинського обласного управління охорони здоров’я
Пілоростеноз (вроджений стеноз воротаря шлунку) – вроджене звуження просвіту пілоричного каналу внаслідок вади розвитку (гіпертрофії і гіперплазії м’язових волокон воротаря шлунку на фоні локального дефіциту, чи дегенеративних змін інтрамуральних нервових клітин).
Етіологія:
Пілоростеноз – захворювання дітей перших місяців життя, викликане звуженням пілоричного каналу внаслідок гіпертрофії м’язових волокон воротаря, його потовщення, головним чином за рахунок м’язових волокон циркулярного шару, яке пов’язують з порушенням інервації, набряком, склерозом слизового і підслизового шару.
Пілоростеноз відноситься до групи багатофакторних захворювань, виникнення яких зумовлене спільною дією генетичних та інших факторів. Всі фактори вказують на полігенне успадкування цієї хвороби з пороговим ефектом.
Етіологія пілоричного стенозу є спірним питанням. Є вирішальні факти, які вказують на генетичне, та такі, що вказують на негенетичне походження даного захворювання. Декілька досліджень показали важливі сімейні акумуляції хвороби.
В багатьох випадках множинних вад, що включають пілоростеноз, відмічені хромосомні (трисомії 18 і 21, часткові трисомії 1 д, 5 д, 13 д; часткові моносомії 15 д і 21 д; та багатогенні синдроми як з аутосомно-домінантним (Альпорта, Пфайффероз), так і з аутосомно-рецесивним успадкуванням (Сміта – Лемлі – Опітца).
Ізольовані форми вродженого пілоростенозу частіше мають мільтифакторіальне походження (Schafer K., Kramer M., 1987 р.).
Частота виникнення сімейних випадків пілоростенозу складає 15%. Емпіричний ризик виникнення повторних випадків в даній сім’ї складає 2-5%.
До цього часу залишаються невиясненими диспропорції частоти захворювань по статі, співвідношення хлопчиків до дівчаток складає 4 до 51, або пілоростеноз коливається від 1 : 150 хлопчиків до 1 : 750 у дівчаток (Вerman R., Vanqnan V., 1987 р.) і, як правило, хворіють першонароджені діти.
Ризик для братів ураженого хлопчика складає 4%, для сестер – 3%, ризик для сибсів враженої дівчинки – відповідно 9% і 4%. Коли пробанд чоловічої статі, то ризик для його синів і дочок відповідно 5,5 – 2,5%, якщо чоловічої статі, то 19 і 7% (Carter C., Evans K., 1969 р.).
При обстеженні внуків та внучок, хворих пілоростенозом, виявилось, що у хворих чоловічої статі ризик для внуків складає 2,3%, для внучок – 0,5%; у хворих жіночої статі – відповідно 4,3 і 1,7% (Carter C. та інш., 1983 р.).
Патогенез:
В даний час існують наступні точки зору на походження пілоростенозу у грудних дітей:
- Bроджене потовщення всіх шарів воротаря.
- Гіпертрофія шарів воротаря є вторинною і виникає на фоні первинного спазму.
- Гіпетрофія пов’язана з недостатністю розвитку іннервації в області воротаря.
Морфологічно при пілоростенозі знаходять надлишковий розвиток сполучної тканини і неправильне розміщення м’язевих волокон з дефіцитом нервових клітин.
Основні критерії, симптоми, ознаки:
Захворювання проявляється на 2-4 тижні, хоча у 5% дітей може проявитись одразу після народження.
Розпочинається із збльовування, яке появляється на 1-2 тижні життя, що переходить у блювоту, яка спостерігається не після кожного годування, але з часом частота і важкість приступів блювоти наростає і набуває вибухового характеру (“вулканом” “гейзером”, “струєю”). Блювота виникає під час годування, зразу ж після годування, а інколи – через декілька годин після годування і носить інтермітуючий характер. Кількість блювотних мас перевищує кількість одноразово з’їдженої їжі (що свідчить про застій в шлунку). Блювотні маси не містять жовчі, а являють собою звернуте материнське молоко з кислим, гострим запахом. Діти постійно неспокійні, голодні, їдять жадно. Стілець появляться рідко і в невеликій кількості, сечовипускання рідші, ніж звичайно.
Тривала повторна блювота приводить до виснаження дитини, зневоднення (сухість шкірних покривів, слизових), метаболічного алкалозу, зниження тургору тканин, зниження температури тіла, артеріального тиску, збільшення гематокриту. Дихання поверхневе, позіхання, сонливість, тремор кінцівок.
При огляді дитини звертають на себе увагу “голодні очі”, як ознака гіпотрофії, хоча апетит збережений.
Після годування у дитини спостерігається здуття епігастральної області і невелике западіння нижнього відділу живота, та перистальтика шлунку у вигляді “пісочного годинника”. Хвиля перистальтики починається від лівого пібребер’я (зліва направо), якщо ці ознаки не надто виражені, то чітко проявляються при постукуванні по животу.
Пропальпувати гіперплазований пілоричний сфінктер можна справа біля краю правого прямого м’язу живота над пупком у вигляді овального щільного утвору, довжиною 2-4 см; найбільш легко пілоричний сфінктер пальпується під час годування після блювоти, або випорожнення шлунка через зонд. При цій маніпуляції не слід застосовувати силу.
В пізніх стадіях хвороби дитина має досить характерний вигляд; бліда, різко зниженого харчування, майже відсутній підшкірно-жировий шар, шкіра суха, збирається в складку, лице зморщене, “старече” з типовими поперечними зморшками на лобі. Стілець диспептичний, “голодний”. Дитина неспокійна, вразлива.
Захворювання ускладнюється частим попаданням значної кількості повітря в шлунок, що спричиняє появу виразок стравохода і шлунка.
Показники обстежень:
Діагноз пілоростенозу встановлюється на основі анамнезу звхворювання, клінічних та рентгеногастрографічних обстежень, фіброгастроскопії, або ультразвукового обстеження.
Для рентгенографії використовують 5% розчин барію в 25-30 мл жіночого молока чи суміш, що вводиться в шлунок через зонд. Рентгенографічні знімки проводять до введення контрасту, через 10-20 хвилин після введення контрасту та через 3-6-24 години.
Рентгенографічні ознаки бувають прямі і непрямі. Прямі характеризують стан просвіту і стінок пілоричного каналу:
- Cимптом “антрального дзьоба” (при введені розчину в шлунок він підходить до пілоричного відділу, відбувається відкриття пілоричного каналу і маса заповняє його початковий відділ, тоді просвіт каналу закривається внаслідок спазму або гіпертрофії м’язів стінок і на рентгенограмі визначається закруглений контур антрального відділу шлунка, що закінчується клиновидним, дзьобоподібним виступом.
- Симптом “вусика жгутика”, що свідчить про звуження та видовження пілоричного каналу.
- Симптом “плечиків”, або “фігурної скобки” – це своєрідний прояв інвагінації, коли гіпертрофовані стінки пілоричного каналу входять у антральний відділ шлунка, або навпаки, стінка антрального відділу насувається на гіпертрофований ригідний воротар.
- Збільшення вираженості складок слизової оболонки шлунка.
Непрямі ознаки пілоростенозу характеризують стан шлунка і кишківника (наявність рідкого вмісту в шлунку натще), мала кількість, або повна відсутність газу в кишківнику, сповільнена евакуація барію з шлунка, мале заповнення, або повна відсутність контрасту 12-палої кишки (через 3-24 год. після введення).
При фіброезофагогастроскопії спостерігається точковий отвір у воротарі, конвергенція складок слизової оболонки антрального відділу шлунку в сторону звуженого воротаря, при інсуфляції повітрям воротар не розкривається, спроба провести ендоскоп у дванадцятипалу кишку неможлива. При проведенні атропінової проби воротар залишається закритим.
Диференціальний діагноз:
- По Б. Братонову (1997 р.) з пілоричним і антральним мембранним стенозом:
- подвоєний воротар;
- hiatus herma;
- анулярна підшлункова залоза;
- пілороспазм, адреногенітальний синдром з втратою солі, сепсис.
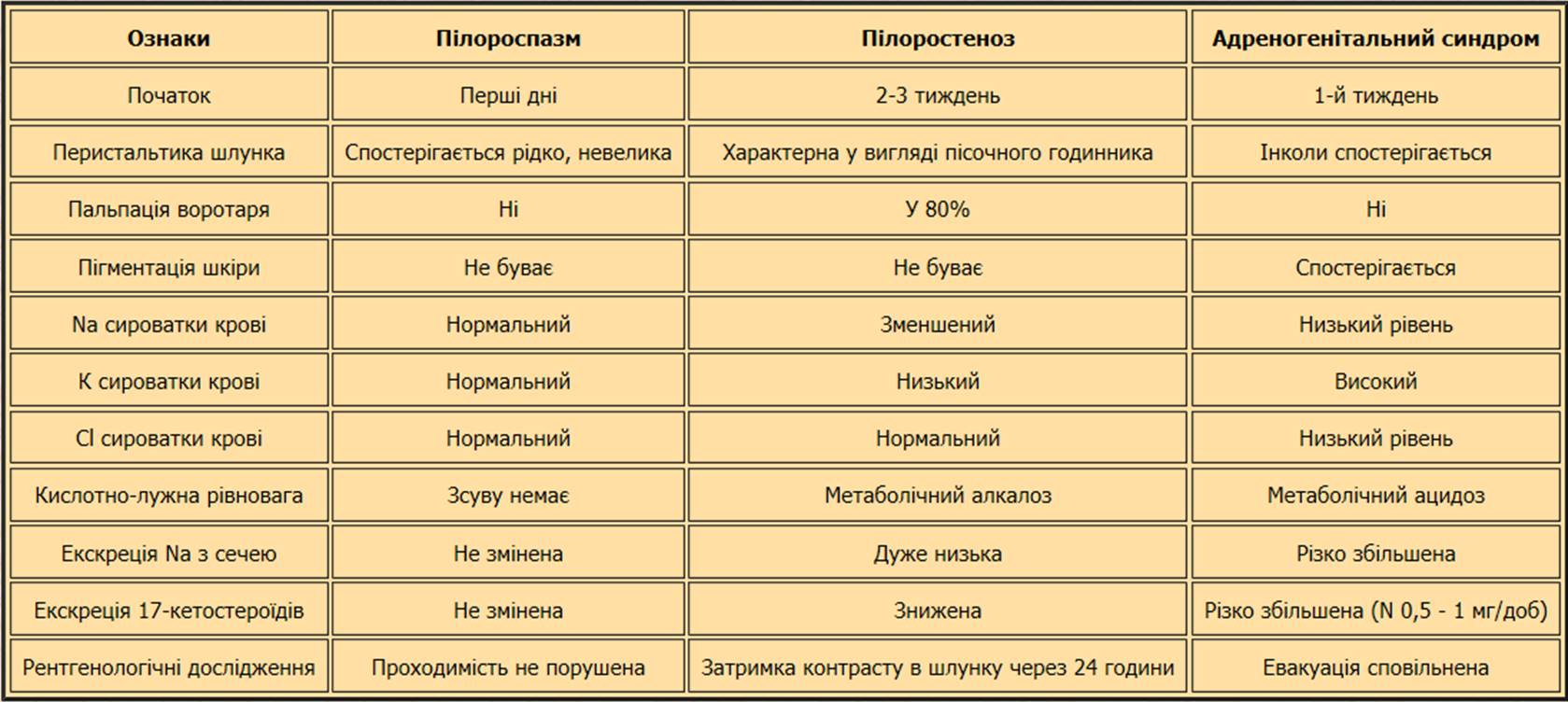
- По Ю.Є. Вельтіщеву з пілороспазмом та адреногенітальним синдромом:
Лікування:
Оперативне – позаслизова пілоротомія по Фреде-Рамштедту чи Баірову, з обов’язковою передопераційною підготовкою від 12 год. до 2 діб з адекватною інфузійною терапією під контролем лабораторних досліджень.
В післяопераційному періоді тактика годування слідуюча: через 2 години по 7-10 мл 5% розчину глюкози per. os. ще через 1 годину 10 мл зціженого грудного молока, а далі через кожні 2 години по 10 мл грудного молока з послідуючою добавкою щоденно по 100 мл грудного молока на добу.
Через 7 діб прикладати до грудей на 7 годувань.
Паралельно продовжується інфузійна терапія з врахуванням об’єму годування та добової потреби рідини.
Профілактика:
Профілактика пілоростенозу не розроблена. Пренатальна діагностика вродженого пілоростенозу особливого значення ні для профілактики, ні для термінів хірургічного втручання немає.
Прогноз:
При своєчасній діагностиці та хірургічному лікуванні благоприємний.
Номер з каталогу МІМ:
179010 Pyloric Stenosis, Infantile.
Література:
- Братанов Б. Клиническая педиатрия.- София, 1989.
- Лазюк Г.И. Тератология человека.- М.: Медицина,1991.
- Пенюшкін А.І. Аномалії розвитку кишківника.- 1984.
- Резнік Б.Я., Мінков І.П. Вроджені пороки розвитку у дітей.- Одеса, 1994.
- Шабалов Н.П. Детские болезни.- Питер, 2000.
Переглянуто редакційною колегією I.B.I.S.: 03/06/2002
Дивіться також:
Рентгенографічні ознаки зрілості скелету (частина 3)
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”
Вік скелету (чоловічий) – 60 місяців
Вік скелету (жіночий) – 47 місяців
Вік скелету (чоловічий) – 84 місяці
Вік скелету (жіночий) – 71 місяць

Вік скелету (чоловічий) – 108 місяців
Вік скелету (жіночий) – 94 місяці

Вік скелету (чоловічий) – 132 місяці
Вік скелету (жіночий) – 113 місяців
Послідовність П’єра Робена
(Pierre Robin Sequence)
В.Л. Терещук
Завідуюча неонатологічним відділенням
Хмельницького пологового будинку
Включення: Синдром П’єра Робена (PRS).
Визначення:
Послідовність П’єра Робена включає:
- мікрогнатію та/або ретрогнатію;
- глоссоптоз та щілину м’якого піднебіння.
Основні діагностичні критерії:
- мала, коротка нижня щелепа (мікрогнатія);
- запала нижня щелепа (ретрогнатія);
- запалий язик (глоссоптоз);
- щілина м’якого піднебіння.
Спостерігається респіраторний дистресс-синдром. Внаслідок обструкції верхніх дихальних шляхів напади апное та ціанозу, які особливо виражені при положенні дитини на спині.
Поєднані симптоми:
У 80% випадків PRS являється складовою частиною іншого синдрому.
Синдроми, асоційовані із послідовністю П’єра Робена:
Моногенні:
- Абрузо-Еріксона синдром;
- Відемана-Беквіта синдром;
- Кампомелічний синдром;
- Цереброкостомандибулярний синдром;
- Вроджена міотонічна дистрофія;
- Діастрофічна дисплазія;
- Дистальний артрогрипоз – послідовність Робена;
- Синдром контрактури – тортіколіз Фростера;
- Ларсена синдром;
- Мандибуло-фаціальний дизостоз;
- Міллера-Діллера синдром;
- Акрофаціальний дизостоз Нагера;
- Ото-палато-дігітальний синдром II;
- Синдром персистуючої лівої верхньої порожнистої вени;
- Синдром колінних птерігіумів;
- Постаксіальний акрофаціальний дизостоз;
- Плечо-ліктьовий синостоз;
- Робена-олігодактилії синдром;
- Вроджена спондилоепіфізарна дисплазія;
- Стіклера синдром;
- Вело-кардіо-фаціальний синдром.
Хромосомні:
- Синдром делеції довгого плеча 4 хромосоми;
- Відемана-Беквіта синдром;
- Синдром делеції довгого плеча 16 хромосоми;
- Синдром дуплікації довгого плеча 11 хромосоми.
Індуковані тератогенами:
- Фетальний алкогольний синдром;
- Фетальний гідантоїновий синдром;
- Фетальний триметадіоновий синдром.
Дизрупції:
Невідомої етіології:
- CHARGE- синдром;
- Синдром дизгенеції стегна – незвичайного обличчя;
- Серединна щілина нижньої губи, піднебіння та гіподонтія;
- Послідовність Мебіуса;
- Асоціація амелія Робен;
- Серповидні лопатки та клишоногість;
- Брюса-Уіншипа синдром.
Ознаки деяких синдромів, асоційованих з PRS
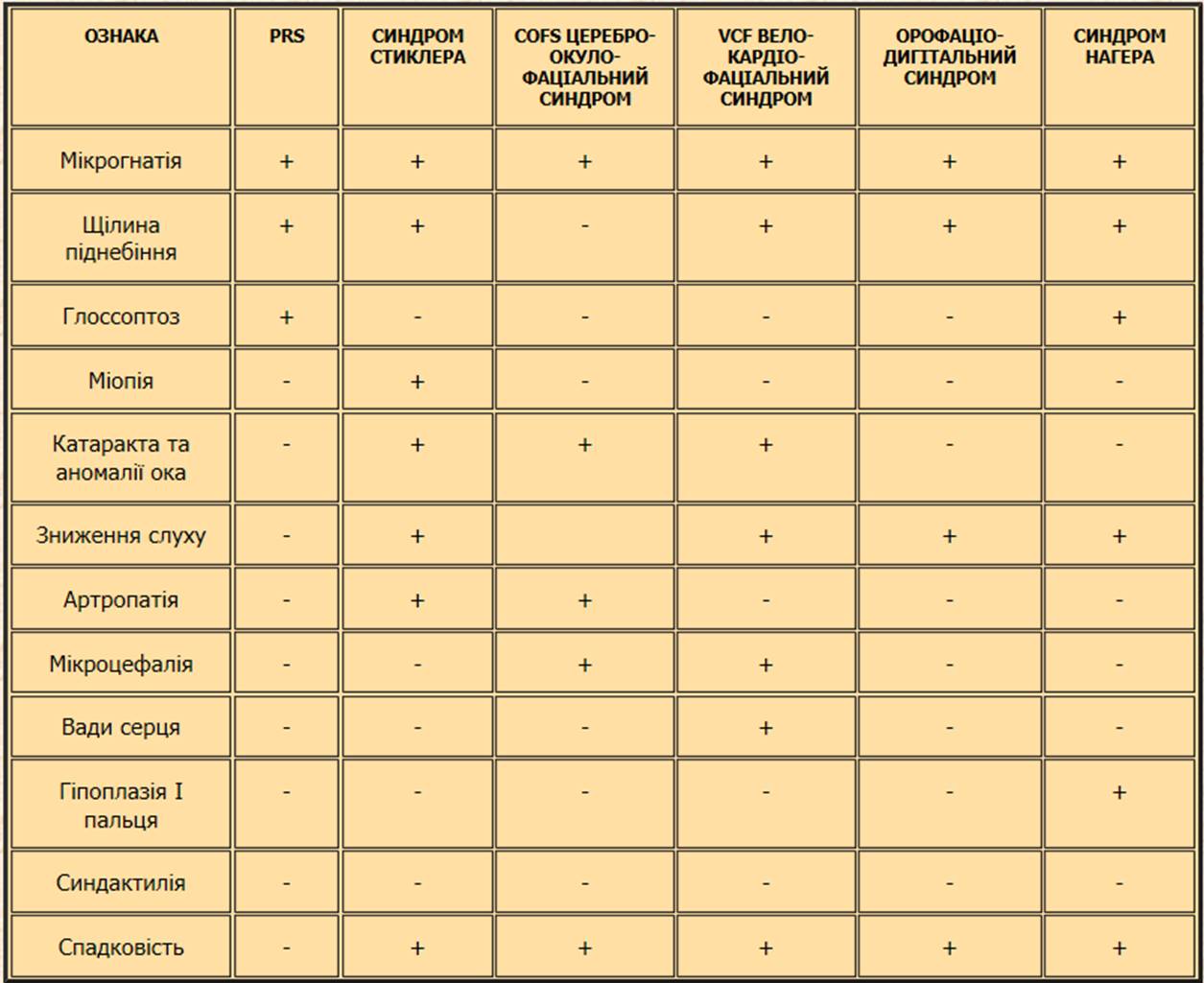 Етіологія і патогенез:
Етіологія і патогенез:
Існуючі гіпотези етіології і патогенезу PRS схематично представлені так:
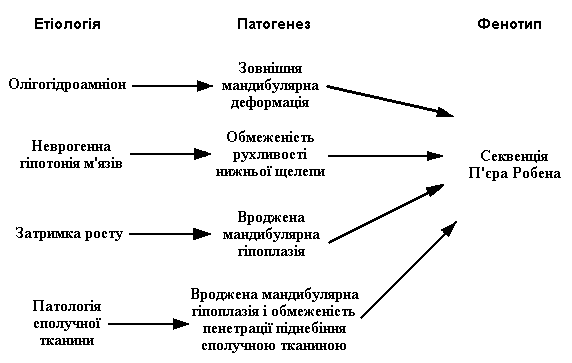
Етіологічна неоднорідність призводить до патогенетичної розрідненості. Розглядаються такі патогенетичні моменти:
Олігогідрамніон приводить до зменшення амніотичної рідини, внаслідок чого виникає надмірне згинання, стискання ембріону, підборіддя надмірно притискається до грудини, що обмежує мандибулярний ріст. Якщо неврогенна гіпотонія обмежує рухливість нижньої щілини раннього фетального періоду до завершення заростання піднебіння, послідовність П’єра Робена може виникнути внаслідок саме мандибулярної обмеженості.
Затримка росту, яка часто супроводжує хромосомні аномалії, наприклад такі, як dup (11q)- синдром, може привести до вродженої мандибулярної гіпоплазії, яка розвивається до 9-го тижня внутрішньоутробного розвитку. Гіпоплазована щелепа зміщує язик дозаду, що порушує процес змикання завіс піднебіння на 8-10 тижні внутрішньоутробного розвитку.
Висловлюється припущення, що глоссоптозу сприяє скорочення підборідно-язикового м’яза. Вроджена недорозвиненість сполучної тканини призводить до дефекту пенетрації піднебіння і виникнення щілини.
Згідно із сучасними уявленнями, первинним фактором порушення дихання у верхніх відділах дихальних шляхів вважається мікрогнатія, а не глоссоптоз.
Частота виникнення: 1 : 8500.
Лікування та нагляд:
Консервативне. Необхідне положення в ліжечку на боці з опущеним головним кінцем. У неонатальному періоді 50% всіх хворих здатні самостійно смоктати і ковтати, інші вимагають годування через назогастральний зонд, або, в деяких випадках, шляхом черезшкірної гастростомії. Для профілактики і лікування дихального дистресс-синдрому застосовується введення повітряпроводу, інтубація трахеї, трахеостомія, фіксація швами верхівки язика до нижніх ясен.
Оперативне:
- Тотальна палаторафія;
- Передня палаторафія;
- Фарингопластика;
- Інтравелопластика;
- Трахеопластика.
При веденні хворих особливої уваги потребує оцінка росту. Порушення фізичного розвитку таких дітей пояснюється труднощами вигодовування, гіпоксемією, дихальною недостатністю, потребою в більшій калорійності харчування, оперативними втручаннями. Крім того, цьому може сприяти супутня патологія, особливо гастроезофагальний рефлюкс та респіраторні інфекції. Затримка розвитку мови проявляється проблемами у вимові фрикативних звуків.
Прогноз:
При адекватному лікуванні та веденні хворого можливе повне відновлення дефіциту зросту та ваги на протязі періоду раннього дитинства. Смертність при PRS коливається від 2,2 до 26% і залежить від віку хворих на момент первинного обстеження та поєднаних симптомів.
Номер МІМ:
261800 Pierre Robin Syndrome.
Література:
- Bixler D, Christian JC. Pierre Robin syndrome occurring in two unrelated sibships. Birth Defects Orig. Art. Ser. 1971;VII(7):67-71.
- Gorlin RJ, Cohen MM Jr., Hennekam RCM. Syndromes of the Head and Neck.- 4th ed.- Oxford: University Press 2001:860-865.
- Houdayer C, Portnoi MF, Vialard F et al. Pierre Robin sequence and interstitial deletion 2q32.3-q33.2. Am. J. Med. Genet. 2001;102:219-226.
- Jones KL. Smith’s Recognizable Patterns of Human Malformation.- 5th ed. Philadelphia: W.B. Saunders.- 1970:234-235.
McKusick VA, Abbey H, Bowen P et al. Medical genetics. 1961. J. Chronic Dis. 1962;15:417-572. - Opitz JM. Personal Communication. Madison, Wis.- 1973.
- Russo G, Mollica F, Pavone L, Musumeci S. Robin’s syndrome in three children of consanguineous parents. A pedigree suggesting autosomal recessive inheritance. Acta Genet. Med. Gemellol. 1973;21:349-353.
- Sachtleben P. Zur Pathogenese und Therapie des Pierre-Robin-Syndroms. Arch. Kinderheilk. 1964;171:55-63.
- Segreti WO, Maumanee P. Familial occurrence of Pierre Robin anomalad. Med. Coll. Va. Quart. 1977;13(4):192-193.
- Shah CV, Pruzansky S, Harris WS. Cardiac malformations with facial clefts. Am. J. Dis. Child. 1970;114:238-244.
- Sheffield LJ, Reiss JA, Strohm K, Gilding M. A genetic follow-up study of 64 patients with the Pierre Robin complex. Am. J. Med. Genet. 1987;28:25-36.
- Singh RP, Jaco NT, Vigna V. Pierre Robin syndrome in siblings. Am. J. Dis. Child. 1970;120:560-561.
- Smith JL, Stowe FR. The Pierre Robin syndrome (glossoptosis, micrognathia, cleft palate). A review of 39 cases with emphasis on associated ocular lesions. Pediatrics 1961;27:128-133.
Переглянуто редакційною колегією I.B.I.S.: 09/09/2002
Рентгенографічні ознаки зрілості скелету (частина 2)
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”
Вік скелету (чоловічий) – 7 місяців
Вік скелету (жіночий) – 6 місяців
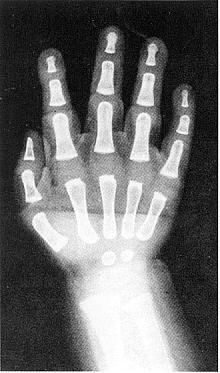
Вік скелету (чоловічий) – 19 місяців
Вік скелету (жіночий) – 14 місяців
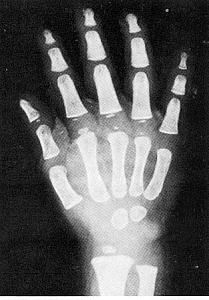
Вік скелету (чоловічий) – 36 місяців
Вік скелету (жіночий) – 27 місяців

Вік скелету (чоловічий) – 48 місяців
Вік скелету (жіночий) – 37 місяців
Паллістера-Холла синдром
(Pallister-Hall syndrome)
Світлана Калинка
Тетяна Христюк
Лікарі-генетики Волинського обласного
дитячого територіального медичного об’єднання
Синоніми:
- Pallister-Hall Syndrome;
- PHS;
- Hypothalamic Hamartoblastoma Syndrome;
- Hypothalamic Hamartoblastoma, Hypopituitarism, Imperforate Anus, and Postaxial Polydactyly.
Основні діагностичні критерії:
- гіпоталамічна гамартобластома, центральна (мезоаксіальна) полідактилія, постаксіальна полідактилія А і В, неперфорований анус;
- розщілина надгортанника, розщілина гортані, аномальна дольчастість легень, агенезія чи дисплазія нирок, вкорочення IV метакарпальних кісток, множинні вуздечки між альвеолярними відростками і слизовою щоки, мікрофтальм, вроджені вади серця (ВВС), гіпоплазія нігтів, затримка внутрішньоутробного розвитку.
Етіологія:
Тип успадкування аутосомно-домінантний. Ризик для сибсів пробанда 50%, якщо захворювання не спричинене спорадичною мутацією.
Проводяться дослідження можливостей діагностики мутацій гена GLI3, який локалізований на хромосомі 7р13.
Клінічна характеристика:
Ріст і розвиток:
- затримка внутрішньоутробного розвитку.
Голова і шия
- Вуха:
- атрезія зовнішнього слухового проходу;
- мікротія;
- диспластичні вуха.
- Очі:
- мікрофтальм.
- Ніс:
- короткий ніс;
- плоске перенісся;
- вивернуті ніздрі.
- Рот:
- множинні вуздечки між альвеолярними відростками і слизовою щоки;
- мікроглосія;
- розщілина губи і піднебіння.
- Зуби:
- зуби новонароджених.
Кардіоваскулярна система
- Серце:
- дефект міжшлуночкової перетинки;
- спільний артеріальний стовбур;
- коарктація аорти.
Респіраторна система
- Гортань:
- розщілина гортані;
- щілина чи гіпоплазія надгортанника (важлива ознака для верифікації діагнозу, так як є рідкісною при інших синдромах, і як ізольована вада).
- Легені:
- аномальна дольчастість легень.
Грудна клітка
- Ребра і грудина:
- злиття ребер.
Шлунково-кишковий тракт
- неперфорований анус
Сечостатева система
- Статеві органи (у чоловіків):
- мікропеніс;
- гіпоплазія статевих органів – вторинна, внаслідок пангіпопітуітаризму.
- Нирки:
- дисплазія нирок;
- ектопія нирок.
Скелетна система
- Спина:
- напівхребці.
- Таз:
- вивих кульшових суглобів.
- Кінцівки:
- дистальне вкорочення кінцівок;
- неповний вивих променевих кісток.
- Кисті:
- центральна полідактилія (мезоаксіальна) – це 6 чи більше добре сформованих пальців з “V-подібними” метакарпальними чи метатарзальними кістками;
- постаксіальна полідактилія А і В;
- олігодактилія;
- синдактилія.
- Стопи:
- синдактилії;
- центральна полідактилія;
- постаксіальна полідактилія А і В.
Шкіра, нігті, волосся
- капілярні гемангіоми середньої частини обличчя;
- гіпоплазія нігтів.
Нервова система
- голопрозенцефалія;
- гіпоталамічна гамартобластома;
- пітуітарна аплазія чи дисплазія.
Гіпоталамічна гамартобластома – це мальформація, а не пухлина. Росте так само, або повільніше мозкової тканини, може досягати величини 4 см, знаходиться в основі заднього ІІІ шлуночка, біля перехрестя зорових нервів і є ізоінтенсивною щодо сірої речовини мозку на Т1 і Т2 при проведенні магнітно-резонансно-томографічного обстеження. Комп’ютерна томографія і нейросонографія не є інформативними. Більшість гамартобластом безсимптомні. Видалення гіпоталамічної гамартобластоми загалом не покращує стан пацієнтів з неврологічною симптоматикою, вони надалі страждають від ятрогенної гіпофізарної недостатності.
Неврологічна симптоматика найчастіше буває у вигляді істеричної епілепсії, яка проявляється клонічними судомами грудної клітки і діафрагми, що імітує сміх; або інших типів судом, які, як правило, резистентні до протисудомної терапії.
Ендокринна система
- пангіпопітуітаризм, що найчастіше проявляється ізольованим дефіцитом гормону росту і передчасним статевим розвитком;
- гіпоплазія наднирників;
- аплазія чи дисплазія щитовидної залози.
Молекулярно-генетичне дослідження:
Відомий лише один ген GLI3, асоційований з синдромом Паллістера-Холла. Діагностика мутацій гена GLI3, таких як 20-23 delG і 20-12 delG доступна для клініцистів. Дослідження інших мутацій гена GLI3 можливе тільки в наукових центрах.
Приблизно половина пацієнтів з синдромом Паллістера-Холла мають мутації гена GLI3.
Діагноз:
- Клінічні ознаки і родинна історія.
- Наявність центральної полідактилії і гіпоталамічної гамартоми є достатньою для постановки діагнозу.
- Легка форма: постаксіальна полідактилія типу А, В, безсимптомна розщілина надгортанника, гіпоталамічна гамартома.
- Важка форма: ларінго-трахеальна розщілина, гіпофізарна недостатність, смерть в періоді новонародженості від недіагностованого адреналового кризу.
- Діагностика мутації гена GLI3, який локалізований на хромосомі 7р13.
Частота:
Рідкісний синдром. На даний час відомо близько 100 сімей з синдромом Паллістера-Холла.
Медико-генетичне консультування:
Співвідношення випадків хвороби, спричинених спорадичною мутацією до випадків, які передались одним із уражених хворобою батьків, невідоме, так як частота незначних ознак синдрому не була ретельно досліджена, і також недостатньо молекулярно-генетичних обстежень.
Достовірність ультразвукового дослідження, як методу пренатальної діагностики, є невідомою. Пренатальне молекулярно-генетичне дослідження можливе.
Генетично-асоційовані захворювання, які пов’язані з мутаціями гена GLI3:
- Грейга цефалосиндактилії синдром (преаксіальна, постаксіальна полідактилія, синдактилія, макроцефалія, гіпертелоризм, антимонголоїдний розріз очей). Проте центральна полідактилія і кісткова синдактилія не є характерними для синдрому Грейга. Більшість пацієнтів з синдромом Грейга мають мутації, що призводять до функціональної гаптонедостатності гена GLI3.
- Ізольована постаксіальна полідактилія тип А.
- Преаксіальна полідактилія тип IV.
Диференціальний діагноз:
З центральною полідактилією:
- МакК’юсік-Кауфмана синдром (центральна чи постаксіальна полідактилія з гідрометрокольпосом і ВВС);
- Холта-Орама синдром (дуже варіабельні аномалії верхніх кінцівок, що можуть включати центральну полідактилію і ВВС);
- Оро-фаціо-дігітальний синдром тип VI (центральна полідактилія з гіпоплазією черв’яка мозочка);
- Барде-Бідля синдром (рідко центральна полідактилія, частіше постаксіальна, ожиріння, дегенерація сітківки);
- Гольцгріва синдром (центральна полідактилія, розщілини піднебіння, ВВС).
З постаксіальною полідактилією:
- Барде-Бідля синдром (постаксіальна полідактилія, ожиріння, пігментний ретиніт, рідко гіпоталамічна гаматробластома);
- Сміта-Лемлі-Опітца синдром (вивернуті ніздрі, шкірна синдактилія пальців стоп, птоз, гіпоспадія і крипторхізм).
Гіпоталамічна гамартобластома:
- Ізольована гіпоталамічна гамартобластома може спричиняти слідуючі ендокринні порушення: ізольований дефіцит гормону росту, передчасний статевий розвиток, істеричну епілепсію, важкі рефрактерні судоми, поведінкові розлади, когнітивну недостатність.
Лікування:
- Діагностика дефіциту наднирників. У немовлят наднирниковий криз може бути летальним, якщо дітям не буде проведено вчасно належне обстеження;
- Визначення секреції гормону росту, фолікулостимулюючого гормону, лютеїнізуючого гормону, гормонів щитовидної залози;
- Рентгенографія кінцівок;
- МРТ головного мозку;
- Ультрасонографія нирок;
- Ларінгоскопія надгортанника, гортано-трахеальної розщілини, якщо пацієнт має симптоми аспірації;
- Обстеження пацієнтів з синдромом Сміта-Лемлі-Опітца;
- Хіругічне лікування атрезії чи стеноза анусу;
- Симптоматичне лікування судом.
Номер з каталогу МІМ:
146510 Pallister-Hall Syndrome; PHS.
Література:
- Biesecker LG. Pallister-Hall Syndrome (GeneReviews).
- Gorlin RJ, Cohen MM, Hennekam RCM. Syndromes of the Head and Neck. Fourth edition. Oxford University Press. 2001:1066-1068.
- Jones KL. Smith’s Recognizable Patterns of Human Malformation. Philadelphia: W.B.Saunders Company 1997:186-187.
Переглянуто редакційною колегією I.B.I.S.: 13/08/2004
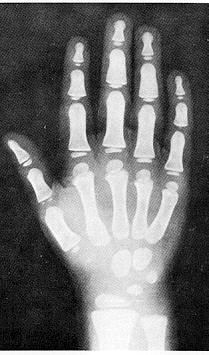
Рентгенографічні ознаки зрілості скелету (частина 1)
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”
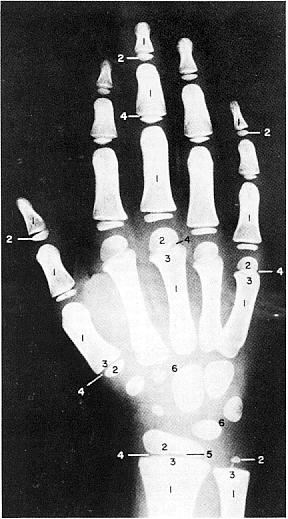
- Діафіз
- Епіфіз
- Метафіз
- Діафізарна ростова хрящова пластинка
- Епіфізарна внутрішня межа кістки та термінальна пластинка
- На знімку не показані суглобний та ростовий хрящовий “обідок” навколо зап’ястку
Вибіркові рентгенограми для визначення кісткового віку подані на наступних сторінках (частини 2, 3 і 4).
Дані отримані від перехресного дослідження 9000 дітей в м. Клівленд, штат Огайо до 1959 року, Pyle SI et al: A Radiographic Standard of Reference for the Growing Hand and Wrist, використовується з дозволу, Bolton-Brush Growth Study Center, Clevelend, Ohio. .
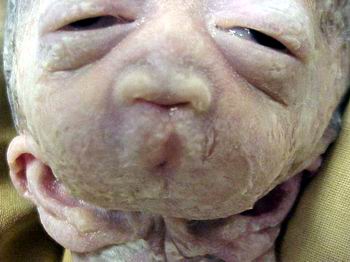
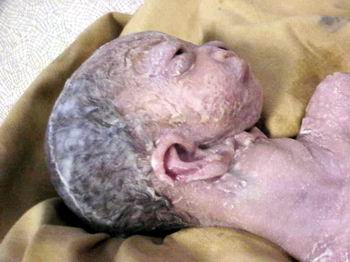
Отоцефалія
(Otocephaly)
Наталія Віталіївна Ткаченко
Лікар акушер-гінеколог
пологового будинку № 2, м. Рівне
Включення:
Синотія, мелотія, агнатія.
Визначення:
Отоцефалія – це вада розвитку, що характеризується потворними змінами рис обличчя, зумовленими відсутністю чи гіпоплазією нижньої щелепи, зближенням скроневих кісток і патологічним горизонтальним розміщенням вушних раковин.
Клінічна картина:
Нижня щелепа відсутня або гіпопластична. Спостерігається вроджена відсутність рота (астомія), але дуже часто є невеликий отвір діаметром 2-3 мм. Спостерігається персистуюча букофарингеальна мембрана і різко виражена мікроглосія. Мальформація зовнішнього вуха. Вуха розміщені вентрально, наближаються або з’єднуються по серединній лінії обличчя. Спостерігається атрезія зовнішніх слухових каналів. В деяких випадках відмічаються аномалії середнього вуха. Дуже рідко відсутні вушні раковини. Може бути розщілина піднебіння.
Інші черепно-лицьові ознаки – це гіпертелоризм, проптоз, блефарофімоз, епібульбарний дермоїд, мікрофтальм, зростання брів – синофриз, розщілина губи, атрезія хоан, аномалії внутрішнього вуха та вестибулярного апарату, роздвоєний надгортанник і нерозвинуті голосові зв’язки.
Асоційовані аномалії:
Змішані аномалії включають в себе гіпоплазію мозочка, septum pellucidum cavum, agenesis of the left calcar avis, зворотнє розміщення органів, однобічну агенезія нирки, ектопію нирок, кисти нирок, агенезію Мюллерівської протоки, крипторхізм, аномалії хребта, відсутність ребер, деформацію Шпренгеля, клишоногість, дефект міжшлуночкової перегородки, стеноз легеневої артерії, відкриту артеріальну протоку, відкрите овальне вікно, відсутність поділу легень на долі.
Ембріогенез:
Нижня щелепа розвивається в результаті злиття двох мандибулярних виступів, які знизу обмежують стомодеум. Первинні зовнішні вуха розміщуються латеральніше і нижче за мандибулярні виступи і в процесі подальшого розвитку зміщуються догори. Вважається, що отоцефалія зумовлена порушенням розвитку нижньої щелепи, можливо, внаслідок дефекту міграції клітин нервового валика. Відсутність чи виражена гіпоплазія нижньої щелепи призводить до патологічного горизонтального розміщення вух з мочками, які розміщені близько до серединної лінії обличчя. Спектр анатомічних порушень при цій патології варіює від грубих змін (розміщення вушних раковин щільно до серединної лінії обличчя: синотія; агнатія і відсутність рота) до порівняно незначних вад (низьке розміщення вушних раковин, мікрогнатія).
Етіологія:
Етіологія невідома. Всі описані випадки ізольованої агнатії були спорадичними. Отоцефалія може бути частиною дуже важких комплексів аномалій, таких як зрощені близнюки, голопрозенцефалія. В експериментальних умовах вада була відтворена у мишей шляхом опромінення рентгенівськими променями. Існує припущення, що отоцефалія може бути спричинена мутаціями в FGF8 (28а).
Диференціальний діагноз:
Необхідно розрізняти ізольовану агнатію та агнатію-голопрозенцефалію. Найбільш виражений випадок – це отоцефалія-циклопія. Так само як і при циклопії, при отоцефалії-циклопії спостерігається алобарна голопрозенцефалія. Причиною цього є дефект прехордальної мезодерми. В літературі описані випадки агнатії-голопрозенцефалії – зворотнього розміщення органів.
Частота:
Максимальна розповсюдженість звичайно менша, ніж 1 на 70000 народжень. Захворювання зустрічається дуже рідко.
Пренатальна діагностика:
Підозра на цю аномалію виникає при неможливості візуалізації нижньої щелепи, низькому розміщенні вушних раковин. Вірогідність отоцефалії велика у плодів з дуже важкими асоційованими аномаліями, такими як аненцефалія, голопрозенцефалія і цефалоцеле. При пренатальному ультразвуковому дослідженні можуть виникнути труднощі при проведенні диференційної діагностики між легкими ступенями отоцефалії та іншими станами, для яких характерне низьке розміщення вушних раковин, наприклад, синдром Трічера-Коллінза. При багатовідді і відсутності ехотіні шлунка плода диференційний діагноз слід проводити з атрезією стравоходу.
Прогноз і акушерська тактика:
Захворювання несумісне з життям. Переривання вагітності необхідно пропонувати при будь-якому терміні вагітності, якщо встановлено заключний діагноз.
Література:
- Пренатальная диагностика врожденных пороков развития плода /Р. Ромеро, Дж. Пилу, Ф. Дженти, А. Гидини, Дж.С. Хоббинс.- М.: Медицина, 1994.- C. 115-117.
- Gorlin RJ, Cohen MM, Hennekam RCM. Syndromes of the Head and Neck. Oxford University Press 2001:717-719.
- Warkany J. Congenital Malformations. Notes and Comments. Year Book Publisher. Chicago 1981:1309, 402.
Переглянуто редакційною колегією I.B.I.S.: 05/01/2004
|
|
|
|
|
|




