
Serhiy
Довжина довгих трубчастих кісток у дівчат (продовження)
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”
Жіноча стать – коливання розмірів трубчастих кісток за даними рентгенографії (в см)
Кістки ноги
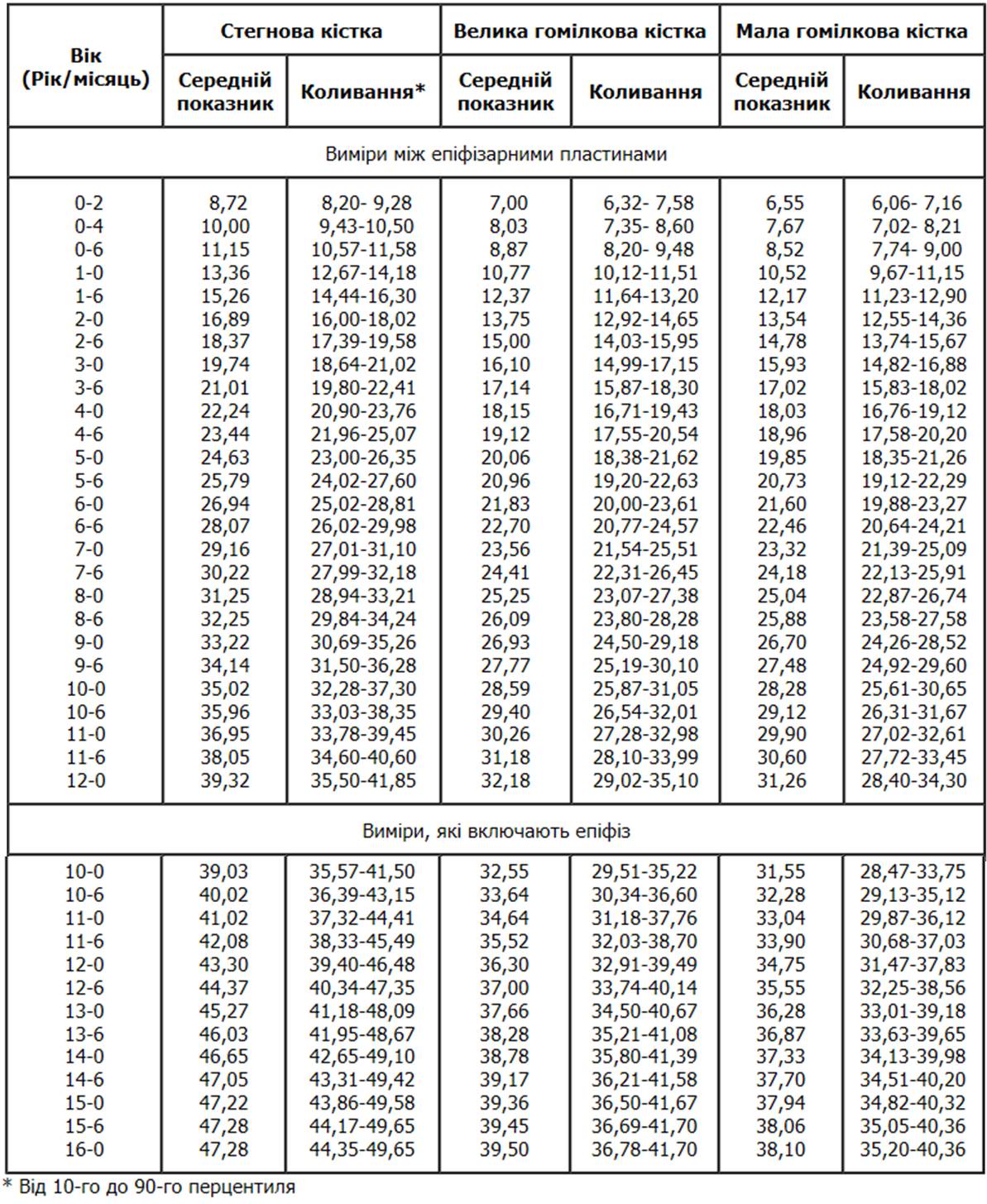
Радіографічні показники, отримані в результаті обстежень 175 осіб віком від 2 місяців до 18 років, які проживали в м. Денвер, штат Колорадо. Усі обстежені були білої раси, за своїм соціальним статусом відносились до середнього класу, походженням переважно з північної Європи. Maresh MM: Am J Dis Child 89:725, 1955.
Остеопетроз
(Osteopetrosis)
Тетяна Краснопера
Спеціаліст з інформаційного забезпечення
Хмельницького ОМНІ-центру
Що таке остеопетроз?
Остеопетроз – це термін, який вживається для опису ряду хвороб, при яких збільшується кісткова маса. При найважчій формі діагноз встановлюється дітям незабаром після народження, в інших же випадках – лише в дорослому віці, часом, як випадкова знахідка при рентгенологічному обстеженні з приводу перелому. Вважається, що всі форми остеопетрозу зумовлені порушенням функції спеціальних клітин кісткової тканини – остеокластів.
Що таке остеокласти?
Протягом всього життя нам необхідно формувати і зміцнювати наші кістки так, щоб вони:
- Залишалися міцними, але легкими, і протистояли тиску і невеликим тріщинам. Щоб максимально корисно використати співвідношення міцності й маси, більшість кісток мають порожнисту або трубчасту структуру.
- Росли разом із дитиною. Це надзвичайно важливо, і є однією з головних проблем дітей із остеопетрозом. Щоб рости і залишатися легкою, кістка всередині повинна мати порожнину і рости з кінців. Ця „конструкція” особливо важлива для кісток черепа, де крізь маленькі отвори нерви проходять до очей, вух, м’язів обличчя, і там, де спинномозкові нерви через нижню частину черепа проходять у хребет. Якщо ці отвори не ростуть так само швидко, як і нерви, то кістки будуть тиснути на нерви, пошкоджуючи їх.
- Містили кістковий мозок. У порожнинах в центрі кісток розташовується кістковий мозок, який є основним виробником крові. Якщо порожнина всередині кістки суттєво зменшується, то кров’яні клітини виробляються переважно в печінці та селезінці – саме такий спосіб кровотворення властивий плоду на ранніх стадіях розвитку. Це спричиняє збільшення печінки і селезінки, що може призвести до руйнування кров’яних клітин і тоді необхідні переливання.
- Підтримували рівні кальцію та фосфатів у крові. Для того, щоб електричні системи, які підтримують роботу наших нервів діяли правильно, рівні кальцію та фосфатів у крові повинні підтримуватися в певних вузьких межах. Наприклад, якщо рівень кальцію зменшується на 25%, то можуть виникнути дратівливість, тремтіння, судоми та тетанія. Надзвичайно важливо, щоб кількість цих елементів чітко контролювалась, оскільки більшість їх запасів міститься саме у кістках.
Всі перераховані цілі досягаються шляхом скоординованої діяльності клітин, які називаються остеокластами та остеобластами. Остеобласти будують кістки, використовуючи кальцій і фосфати (разом із іншими речовинами) у якості будівельних блоків. Їхня активність урівноважується остеокластами, великими клітинами, утвореними внаслідок злиття декількох менших клітин. Результатом такого злиття є утворення у клітині множинних ядер (центральної частини клітини, де містяться гени).
Остеокласти утворюються із клітин, які продукуються кістковим мозком і переміщуються до кістки – ось чому трансплантація кісткового мозку використовується при лікуванні важких форм цього захворювання у дітей. Сформовані, вони міцно прикріплюються до ділянки кістки і вивільняють речовини, які “розчиняють” кістки.
Однією з речовин, яка “розчиняє” кістки, є соляна кислота. Вона утворюється з іонів водню та хлориду, які відкачуються з остеокластів на поверхню кісток крізь іонні канали.
Доведено, що приблизно у половині випадків остеопетрозу причиною цього захворювання є мутації генів, які відповідають за генетичний код іонних каналів.
Поверхня остеокластів – хвиляста (як, скажімо, поверхня тонкого кишківника), щоб максимально збільшити площу поверхні, яка контактує з кісткою. Це називається хвилястою межею. Коли ділянка кістки розчинена, остеокласти “виповзають” із утвореного ними заглиблення на нову ділянку кістки.
При остеопетрозі остеокласти або відсутні взагалі, або не здатні утворювати хвилясті межі, або збільшуються у кількості. Останнє дуже часто зустрічається у дітей із важкими формами. Можливо, організм розпізнає дефектні остеокласти, як недіючі, і таким чином ”наказує” їм множитись, в інших випадках остеокласти просто не сформовані.
Які є типи остеопетрозу?
Описані різні форми захворювання, нижче вони перераховані у порядку зростання важкості.
- Транзиторний інфантильний остеопетроз. Інформації про дану форму захворювання – яка зникає без втручання – найменше. Вона є легкою формою захворювання і може виявлятися у носіїв у ранньому віці, хоча це і не доведено. Про цю форму важливо знати, бо вона вказує на необхідність рентгенівської перевірки всіх дітей, яким будуть трансплантувати кістковий мозок перед застосуванням хіміотерапії. Це знизить ризик (хоч він і незначний) пересадки мозку, хворого на транзиторний інфантильний остепетроз.
- Дорослий, „доброякісний”, аутосомно–домінантний остеопетроз. Це перша описана (німецьким рентгенологом Альберс-Шонбергом) форма захворювання, яка трапляється у двох формах:
- І тип – для нього типовим є потовщення (склероз) верхньої частини черепа (склепіння черепа);
- ІІ тип – (класична хвороба Альберс-Шонберга) при цьому типі хребет має вигляд так званого „зім’ятого джерсі” та тазові кістки містять “кістку в кістці”.
Це найбільш розповсюджена форма захворювання. Діагноз зазвичай встановлюється в дорослому або підлітковому віці. За різними оцінками уражається 1 з 20000–50000. Тип успадкування – аутосомно–домінантний, хоча часто пенетрантність змінюється. Найчастіше зустрічаються такі симптоми, як підвищений ризик переломів, болі в спині, кістках, головні болі та остеомієліт (інфекційне кісткове захворювання). Проблеми внаслідок стиснення нервів (глухота, втрата зору, параліч лицьових нервів) зустрічаються набагато рідше, ніж при важких формах інфантильного остеопетрозу.
Ген, відповідальний за ІІ тип захворювання, розташований на хромосомі 1 (абревіатура 1р21), як було точно вказано Вім ван Халом та його колегами у Бельгії.
- Дефіцит карбоангідрази ІІ типу (САІІ). Хвороба викликається дефіцитом ферменту САІІ, який діє у кістках, нирках і мозку, тому уражаються всі ці органи. Хвороба рідкісна, найчастіше нею страждають діти середземноморських та арабських країн. Ген, відповідальний за виробництво САІІ розташований на 8 хромосомі (8q22). Це єдина форма захворювання, яка діагностується в ранньому дитинстві шляхом ідентифікації гена, що зумовлює хворобу.
Окрім збільшення кісткової маси та схильності до переломів, відбуваються також і характерні зміни хімічного складу організму. Кров злегка кисла, концентрація хлориду в ній висока (гіперхлоремічний ацидоз). Кислота крові викликана надмірною втратою бікарбонату із ниркових канальців (нирковий канальцевий ацидоз).
САІІ відіграє важливу роль і у функціонуванні мозку, тому в уражених дітей розвивається прогресуюча церебральна кальцифікація та розумова відсталість. Іншими проблемними моментами є затримка росту та неправильний прикус тощо.
Симптоми виникають, як правило, в перші роки життя, хоча рентгенографічних змін при народженні не відзначається. Більше того, протягом життя рентгенівська картина може поліпшуватися. Приблизно у 3/5 хворих дітей розвиваються симптоми стиснення черепних нервів: найчастіше це викликає сліпоту, але також може призвести до втрати слуху та паралічу лицьових мязів. На відміну від злоякісного інфантильного остеопетрозу, який описано нижче, проблеми із кров’ю зазвичай незначні, чи відсутні взагалі.
Лікування. Пероральний прийомом бікарбонатів не впливає на перебіг захворювання. Трансплантація кісткового мозку нормалізує кісткову масу, виробляючи здорові остеокласти. Наразі ще не відомо, чи відверне це церебральну кальцифікацію та розумову затримку, хоча це навряд чи можливо.
Цю хворобу необхідно виключити у кожному випадку захворювання дитини на остеопетроз, принаймні шляхом уважного дослідження бікарбонату та хлориду крові (рН сечі, але бажано вимірювання активності САІІ). Особливо це важливо для дітей середземноморських та арабських країн та для дітей із захворюваннями крові.
- Злоякісний інфантильний остеопетроз. Це найбільш розповсюджена складна форма інфантильного остеопетрозу, яка, ймовірно, викликана дефектами декількох генів, оскільки не було визначено окремого гена (локуса). У більшості дітей розвиваються серйозні нервові ураження в ранньому дитинстві і без лікування 2/3 хворих дітей помирають до шестирічного віку. Наразі єдиним дієвим методом лікування є трансплатація кісткового мозку. Це може повністю запобігти подальшому прогресуванню хвороби.
Найбільш розповсюдженими симптомами є (у порядку частоти виникнення):
- Нежить (застуда). Закупорка носового каналу та виділення починаються з раннього віку і, зазвичай, не припиняються. Це викликано надмірним ростом верхньої носової кістки, а не постійними вірусними інфекціями, як дехто вважає.
- Слабкий зір. Це найбільш частий симптом, який є приводом до діагностування. Він стає очевидним на 2–4 місяці життя. Приблизно у 75% дітей, хворих на злоякісний інфантильний остеопетроз, слабне зір (переважно на першому році життя). У багатьох випадках пошкодження зорового нерву стає очевидним вже у 6-8 тижнів, коли, як правило, діти починають фіксувати погляд на обличчях, плямах світла та великих предметах і стежити за ними. У зв’язку із відсутністю належної фіксації, очі починають швидко рухатися зі сторони в сторону. Ці рухи називаються ністагмом.
Зоровий нерв передає імпульси від сітківки, яка розташована позаду ока, до мозку, проходячи на своєму шляху через кістковий зоровий канал у черепі. Якщо на цій ділянці остеокласти не протидіють утворенню остеобластами нової кістки, то зоровий канал швидко звужується і тисне на кров’яні судини, які оточують і живлять нерв. Від нестачі крові нерв відмирає і тоншає (атрофія зорового нерву), а згодом може зовсім зникнути. Це можна побачити на томограмі.
У багатьох дітей пошкодження зорового нерва відбувається ще в утробі матері, хоча це може і не проявитися одразу. Як і у випадках інших пошкоджень нервів, цей процес є незворотним, отож, якщо зір втрачено – його не повернути (навіть після успішної трансплантації кісткового мозку). Лікар може побачити пошкодження нерва, обстежуючи сітківку офтальмоскопом там, де головка нерва (оптичний диск) виглядає дуже блідою. Дитині, яка тільки починає сліпнути, врятувати зір можливо за допомогою хірургічної операції, яка називається “декомпресія зорового нерва”.
У окремих пацієнтів може розвинутись переродження сітківки, зазвичай, у поєднанні із важкою нейродегенеративною хворобою.
- „Великі голови”. Потовщення кісток у основі черепа набагато серйозніше, ніж у склепінні. Це спричиняє значну опуклість лоба, а зовній вигляд погіршується через поганий ріст інших частин тіла – діти з остеопетрозом відносно низькорослі, і як наслідок цього, їх зросто–вагові криві нижчі за норму.
- Плаксивість/дратівливість і переломи. Плаксивість, на відміну від нежиті, не завжди включається до переліку головних симптомів. Між тим, часто діти з остеопетрозом дратівливі, їх важко заспокоїти. Вважається, що головним чином це пов’язано із недіагностованими переломами і головними болями в результаті підвищеного внутрішньочерепного венозного тиску, причиною якого є стиснення у яремному отворі однієї з головних вен, яка відводить кров від мозку.
У більшості дітей, при ретельному дослідженні рентгенівських знімків їх кісток, знаходять переломи. Особливо часто ламаються довгі кістки кінцівок, ребра і акроміальні відростки (невеличкі кісткові відростки від лопатки). Очевидно, вони виникають під час проходження дитини родовим шляхом, оскільки кістки хворого на остеопетроз дуже крихкі (нагадують брусочки крейди). Ці переломи викликають біль, особливо, коли дитину брати на руки, тому хворі діти дуже погано їдять. Тепер на ранніх етапах використовують знеболюючі препарати.
- Симптоми низького вмісту кальцію (нервове тремтіння/напади). На першому місяці життя дитина знаходиться на дуже важливому етапі стосовно балансу кальцію в організмі через швидку мінералізацію і ріст кісток, це може також погіршуватися і незбалансованим харчуванням. Якщо рівень кальцію знижується дуже сильно, дитина стає дратівливою і тремтить нервово. У найгіршому випадку, у дітей можуть початися напади і остеопетроз вважають потенційною причиною неонатальних конвульсій, які виникають на першому місяці життя. Визнання цього зв’язку є дуже важливим, оскільки встановлення діагнозу в цьому віці може допомогти шляхом трансплантації врятувати дитині зір.
- Захворювання крові (анемія/тромбоцитопенія). Кров містить чотири основних типи клітин:
- червоні кров’яні клітини, які розносять кисень по тілу, їх нестача називається анемією;
- тромбоцити – маленькі клітини, які прикріпляються до пошкоджених частин кров’яних судин; якщо їх кількість зменшується, то на шкірі часто виникають синці та петехіальні крововиливи. Нестача тромбоцитів називається тромбоцитопенією і може призвести до серйозних кровотеч;
- лейкоцити чи нейтрофіли – білі кров’яні клітини, головна мета яких – боротися із хворобами. Гній переважно утворюється мертвими лейкоцитами. Їх нестача викликає нейтропенію;
- лімфоцити – інший тип білих кров’яних клітин, які допомагають боротися із вірусними інфекціями та виробляти антитіла.
На ранній стадії внутрішньоматкового розвитку кров головним чином виробляється печінкою і селезінкою. В другому триместрі вагітності ця функція поступово переходить до кісткового мозку, і до народження дитини формується повністю. Якщо цього не відбувається, то печінка і селезінка залишаються великими (гепатоспленомегалія), їх легко промацати у черевній порожнині. Якщо розглядати кров під мікроскопом, то вона має характерні особливості, які залежать від місця її утворення (лейкоеритробластична анемія). Високим є вміст білих кровяних клітин і стовбурових клітин – попередників усіх перерахованих вище клітин. Часто зустрічаються анемія та низький рівень тромбоцитів, можливо, внаслідок підвищеного рівня руйнування клітин крові селезінкою. З віком стан погіршується, оскільки селезінка збільшується у розмірі. Врешті–решт, у зв’язку із гіперспленізмом/спленомегалією, життєво важливими стають постійні переливання крові та тромбоцитів.
У близько 3/4 дітей протягом першого року життя виникають проблеми із кровоносною системою. Зрідка відбувається нормалізація розмірів печінки та селезінки, зникають симптоми захворювання крові, хоча щільність кісток залишається високою. Хворі діти, у яких проблеми з кров’ю розвиваються протягом перших 3 місяців життя, мають невелику перспективу.
- Інфекції. Схильність до інфекційних захворювань може виникнути через низький вміст білих тілець у крові, викликаний гіперспленізмом. Проте, очевидним є також послаблення механізму знищення бактерій нейтрофільними лейкоцитами. Нейтрофільні лейкоцити, або нейтрофіли, виробляють дуже токсичні пероксиди, які використовуються для знищення мікробів. Це є основою лікування, яке базується на внутрішньому прийомі гамма-інтерферону, який знижує рівень інфікування. Інфекції більш розповсюджені у старших дітей, особливо часто зустрічається остеомієліт (інфекційне захворювання кісток). Пацієнти з обома формами захворювання – інфантильним та дорослим остеопетрозом – часто страждають від уповільненого прорізування зубів та їх поганої якості – зуби дуже швидко псуються, і це створює передумови для виникнення серйозних форм остеомієліту щелеп та кісток.
- Нейронопатичний остеопетроз. Найбільш складна форма остеопетрозу, позаяк набуває ознак нейро-дегенеративного захворювання, протягом перших місяців життя у хворих дітей виникає спастичність.
Багато дітей із нейронопатичним остеопетрозом сліпі від народження, мають складні розлади системи кровообігу і помирають протягом першого року життя. Біопсія головного та спинного мозку показує наявність “включених тілець”, схожих на ті, які зустрічаються при ряді невиліковних нейродегенеративних хвороб, цероїдному ліпофусцинозі, краще відомому як хвороба Баттена. У деяких дітей із цією формою захворювання може бути також і ретинопатія: дегенеративна хвороба сітківки ока, яку можна визначити при офтальмологічному огляді.
На жаль, дуже важко відрізнити дитину з таким захворюванням від дитини із злоякісним інфантильним остеопетрозом. У цьому можуть допомогти дослідження: на комп’ютерній томограмі мозку можна побачити церебральну атрофію, а на електроенцефалограмі (запис імпульсів мозку) – певні відхилення. Проте, у дітей із цією формою хвороби всі ці показники можуть бути в нормі. На жаль, біопсія нервової тканини часто не дає результатів через низький вміст тромбоцитів, і в такому випадку необхідними є біохімічні/ генетичні засоби діагностування.
Лікування злоякісного інфантильного остеопетрозу
Які перспективи, якщо не лікувати?
За відсутності лікування лише 30% дітей із злоякісним інфантильним остеопетрозом доживають до 6-річного віку, і практично всі помирають ще у підлітковому віці. Якість їхнього життя часто також низька, багато з цих дітей сліпі, а пізніше втрачають і слух, у них підвищується тиск спинномозкової рідини і розвивається гідроцефалія (водянка головного мозку). В свою чергу це призводить до тиску на мозок, і, як наслідок, до розумової відсталості.
За відсутності лікування лише 30% дітей із злоякісним інфантильним остеопетрозом доживають до 6-річного віку, і практично всі помирають ще у підлітковому віці. Якість їхнього життя часто також низька, багато з цих дітей сліпі, а пізніше втрачають і слух, у них підвищується тиск спинномозкової рідини і розвивається гідроцефалія (водянка головного мозку). В свою чергу це призводить до тиску на мозок, і, як наслідок, до розумової відсталості.
З часом здатність пацієнта відкашлювати мокротиння знижується і у багатьох розвиваються серйозні інфекційні захворювання легень. Прогресуючий гіперспленізм та зниження вмісту кров’яних тілець може ускладнити цей процес.
Які є способи лікування?
Є два основних способи лікування: а) медикаментозна терапія, б) трансплантація кісткового мозку. Ліки використовують для того, щоб полегшити симптоми захворювання, тоді як трансплантацію кісткового мозку – для того, щоб вилікувати хворобу. При цьому, хірургічне втручання може бути двох видів: для тих, хто швидко втрачає зір, може бути запропонована операція декомпресії зорового нерва; при гідроцефалії стан полегшується шляхом вживлення шунту.
Медикаментозне лікування. Основними препаратами, які використовуються, є:
- Кальцитріол (1,25- гідроксид вітаміну D). Ці ліки призначалися у великих дозах у зв’язку із припущенням, що остеокласти при злоякісному інфантильному остеопетрозі можуть бути стійкими до стимулюючого ефекту звичайної дози вітаміну D. Було описано лікування 2 пацієнтів. У першого збільшилась кількість “хвилястих меж” на остеокластах, підвищення вмісту хімічних речовин у крові та сечі свідчили про посилення діяльності остеокластів. Проте у клінічній картині значного поліпшення не спостерігалося. У другого пацієнта не було покращення ні в остеокластах, ні у виділенні кальцію. Це лікування більше не використовується.
- Преднізолон. Ці стероїдні ліки призначалися як у великих, так і у малих дозах. Основним позитивним результатом є вплив на збільшену селезінку і полегшення симптомів анемії та тромбоцитопенії. При високих дозах може відбуватися зниження щільності кісток та розміру порожнин кісткового мозку, вірогідно це пов’язано із потоншуючим впливом стероїдів. Між тим, ці дії нетривалі, а високі дози гормонів мають багато несприятливих побічних ефектів: схильність до частих інфекційних захворювань (особливо молочниця ротової порожнини), збільшення ваги, набряки обличчя, діабет, високий кров’яний тиск, а з часом розвивається катаракта. Низькі дози стероїдів мають меншу кількість побічних ефектів, хоча можуть суттєво знизити потребу в переливаннях. Про таке лікування варто серйозно подумати, якщо хвора дитина не є кандидатом на пересадку кісткового мозку.
- Інтерферон-y (гамма) 1b. Використання інтерферону-y (гамма) обумовлено тим, що у багатьох пацієнтів із остеопетрозом виявляється відхилення від норми у системі пероксидів, які використовуються нейтрофілами для знищення бактерій. Це викликає певне зацікавлення, оскільки і остеокласти, і нейтрофіли виробляються тими ж самими клітинами – попередниками у кістковому мозку, а також утворення пероксидів вірогідно має значення при розчиненні кістки остеокластами. Звідси логічним є використання інтерферону-y (гамма) – ліків, які можуть стимулювати діяльність пероксидів.
Перед обговоренням результатів терапії інтерфероном-y (гамма), необхідно наголосити, що лише при інфантильному остеопетрозі функція пероксидів відхиляється від норми відповідно до популяції нейтрофілів. Цим пацієнти із інфантильним остеопетрозом відрізняються від хворих із хронічною гранулематозною хворобою (ХГХ), у яких відхилення функції пероксидів відбувається у всій популяції нейтрофілів. У хворих на ХГХ розвиваються часті важкі хронічні бактеріальні та грибкові інфекції, які можуть бути значно полегшені лікуванням інтерфероном-y (гамма). Для порівняння, хворі на остеопетроз справді часто хворіють інфекційними хворобами, особливо кісток щелепи (як можливий наслідок порушеного росту зубів та карієсу), проте вони не є такими характерними ознаками як при ХГХ. У деяких випадках не відзначається підвищення рівня інфекційних захворювань у порівнянні із здоровими дітьми, принаймні у ранньому віці.
Інтерферон-y (гамма) 1b вводиться підшкірно тричі на тиждень. Наразі повідомлень про серйозні побічні дії немає. Використання цих ліків було описано на прикладі 14 хворих дітей, 11 з них отримували ліки протягом принаймні 18 місяців. У всіх пацієнтів спостерігалося розсмоктування кісток та збільшення порожнин кісткового мозку. Крім того, повідомлялося про значне зниження рівня інфекційних хвороб. Ця терапія пройшла широкі випробування, особливо у США, оскільки вона набагато безпечніша, ніж пересадка кісткового мозку. Між тим, деякі родини побажали припинити лікування у зв’язку із необхідністю робити постійні ін’єкції та через те, що хвороба не виліковується, а лише поліпшується стан хворого. Терапія інтерфероном-y (гамма) 1b особливо важлива у випадках, коли необхідний для пересадки кісткового мозку донор відсутній, навіть при тому, що із поліпшенням результатів пересадки від „частково придатних” донорів, зменшиться кількість показів до такої терапії.
Хірургічне втручання. Однією із хірургічних операцій при остеопетрозі є вживлення шунта, що застосовується для лікування гідроцефалії, розширення шлуночків у глибині головного мозку. Гідроцефалія спричинюється надлишковим ростом в основі черепа, що заважає нормальному руху спинно-мозкової рідини (СМР). Такі симптоми як дратівливість та періодична рвота можуть і не проявитися. Нелікована протягом довгого часу гідроцефалія призводить до затримки розумового розвитку.
Надлишковий тиск може бути зменшений шляхом вживлення шунта – трубки, яка вставляється в один із шлуночків головного мозку і ведеться під шкірою до черевної порожнини. Рідина таким чином витікає із шлуночка до черевної порожнини, де і перепоглинається. Зазвичай, шунт має односторонній клапан, який можна промацати за вухом; це забезпечує захист від перетікання рідини із черевної порожнини у шлуночок головного мозку, а також дозволяє оцінити тиск на головний мозок (оскільки шунти можуть бути заблоковані, особливо у нижній частині). Завдяки клапану є можливість взяти зразок спинномозкової рідини, якщо виникають підозри про інфікування шунта (саме це і є найбільш серйозним ускладненням при вживленні шунта).
Генетика остеопетрозу
Остеопетроз успадковується двома шляхами: за аутосомно-рецесивним та аутосомно-домінантним типом.
Генетична інформація клітини міститься у клітинному ядрі, в якому знаходяться хромосоми, утворені згорнутою в спіраль ДНК, що в свою чергу є ланцюжком генів, кожен з яких кодує певний білок. Якщо у гені трапляється зміна, яка називається мутацією, вона діє як помилка у „правописі” ДНК – змінюється склад білка, або шлях його утворення. Деякі зміни у генах успадковуються від батьків, інші виникають у певної дитини вперше (іноді цю зміну називають “de novo”). Якщо мутація виникла у дитини вперше, то шанс появи такої ж мутації у її сестри чи брата дуже незначний. Проте, вірогідно, що цей ген передасться нащадкам пацієнта.
Аутосомно–рецесивне успадкування. При цьому типі успадкування для розвитку хвороби необхідно, щоб обидві копії певного гену (тобто материнська і батьківська копії) змінились, мали мутацію.
У кожного із батьків, від яких успадковується ген, є лише по одній “дефектній копії”. Це забезпечує утворення відповідного білка наполовину – зазвичай, цього більш ніж достатньо для нормального функціонування, – і симптоми хвороби відсутні. При цьому батьки можуть передавати свій ген дітям – таким чином обоє батьків називаються носіями.
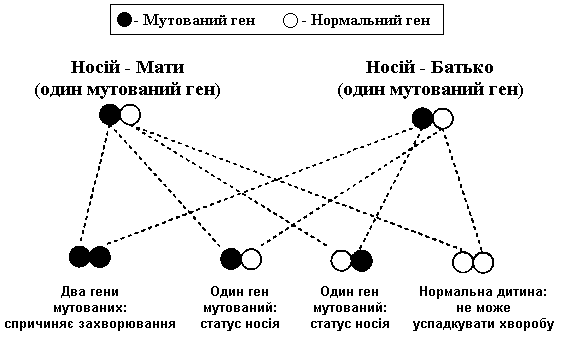
Процес розподілу хромосом в яєчниках і спермі надзвичайно випадковий. Статистика показує, що 25% дітей народяться хворими, 25% – здоровими, а 50% будуть носіями, як і їх батьки. Проте ситуацію передбачити неможливо. Так, як і у лотереї – шанси виграти однакові кожного разу, незалежно від того скільки вже раз ви грали. Так і з успадкуванням гену. Якщо у пари носіїв дефектного гену 4 дітей, то всі вони можуть бути уражені, або не уражені – це випадковість. Тільки якщо у них дуже багато дітей, то тоді статистика вирівнюється: якщо б у них було 400 дітей, то вірогідно, що 100 з них були б хворими.
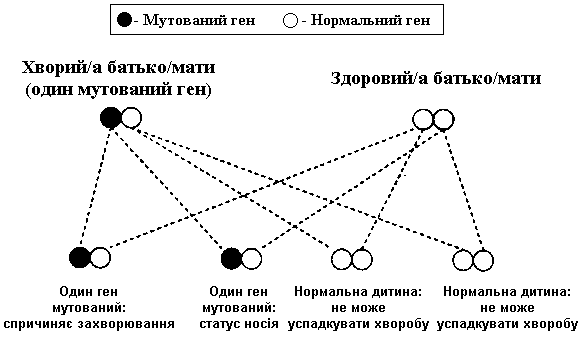
Аутосомно-домінантні захворювання. Іноді дефектний білок, що виробляється однією копією аутосомного гена, діє так сильно, що спричинює хворобу, незважаючи на те, що виробляється клітинами, де активною є інша копія гена. Цей тип захворювання називається домінантним, і характеризується успадкуванням лише від одного з батьків, який хворіє сам. Однак, основні метаболічні ланцюги людського організму складні, багатокомпонентні, та різноманітні у комбінаціях своїх елементів. Зазвичай це означає, що хвороба виникає менш серйозна, ніж це передбачалося її генетикою. Наприклад, у батька (або матері) форма захворювання серйозна, тоді як дитина може бути уражена набагато легше. Це називається варіативним успадкуванням, хоча механізми, відповідальні за цей процес, переважно є ще невідомими.
Переглянуто редакційною колегією I.B.I.S.: 04/11/2003
Дивіться також:
- Остеопетроз (інформація для спеціалістів)
Довжина довгих трубчастих кісток у дівчат
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”
Жіноча стать – коливання розмірів трубчастих кісток за даними рентгенографії (в см)
Кістки руки
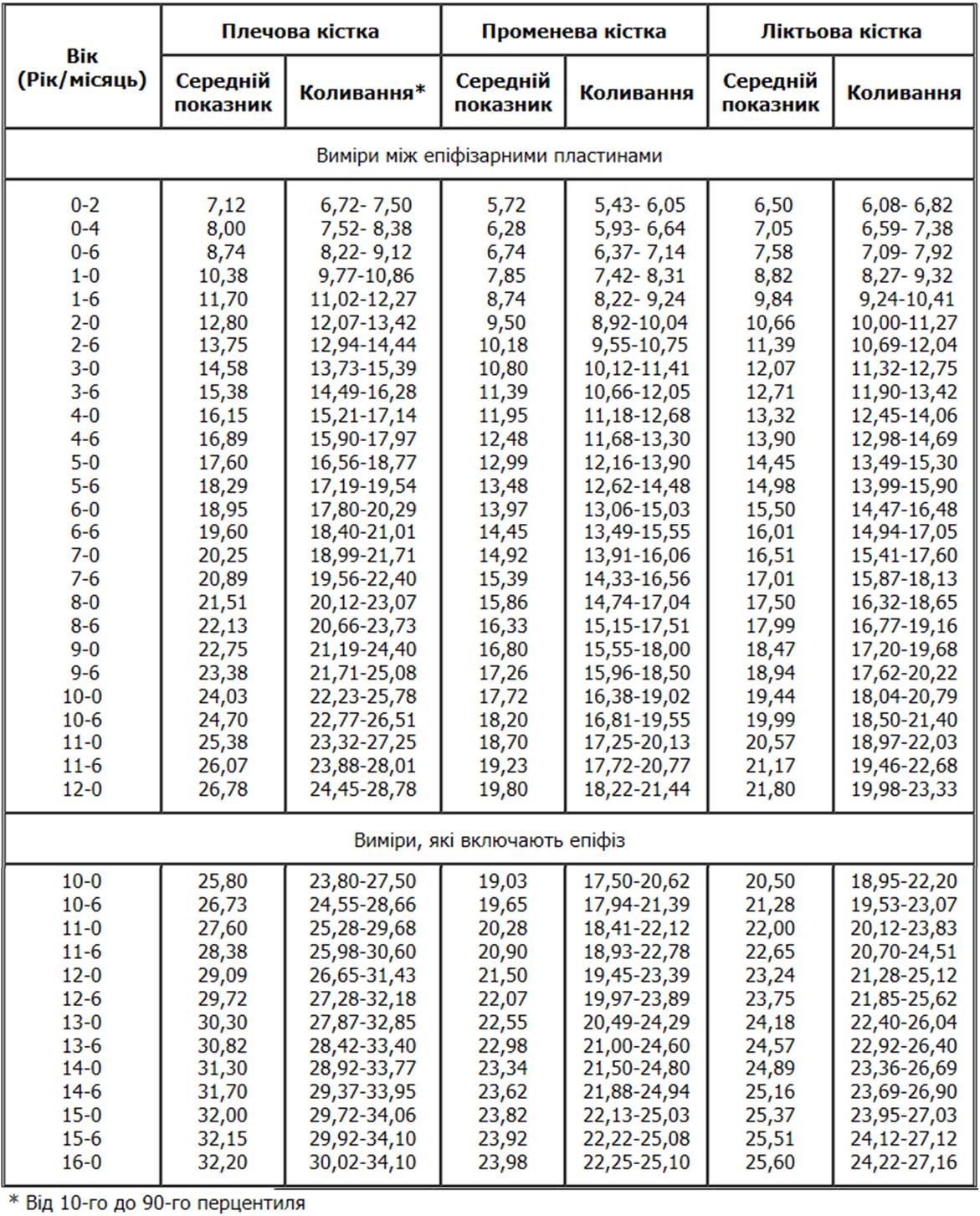
Радіографічні показники, отримані в результаті обстежень 175 осіб віком від 2 місяців до 18 років, які проживали в м. Денвер, штат Колорадо. Усі обстежені були білої раси, за своїм соціальним статусом відносились до середнього класу, походженням переважно з північної Європи. Maresh MM: Am J Dis Child 89:725, 1955.
Остеопетроз
(Osteopetrosis)
Наталія Зимак-Закутня
Завідуюча Хмельницькою
медико-генетичною консультацією
Остеопетроз – це група спадкових захворювань, об’єднаних спільним патогенетичним механізмом (порушенням процесів резорбції кісток остеобластами), що призводить до збільшення кісткової маси.
Перший опис захворювання належить німецькому рентгенологу Альберсу-Шонбергу (Albers-Schonberg, 1904 рік).
Синоніми:
Хвороба Albers-Schonberg, хвороба мармурових кісток, остеокластична кісткова резорбція.
Етіологія та патогенез:
Первинним дефектом для усіх форм остеопетрозу є порушення резорбтивної функції остеокластів, що приводить до збільшення склерозованої кісткової маси, яка має знижені механічні властивості. Молекулярні механізми та сайти мутацій здебільшого остаточно не з’ясовані, за винятком генетичного дефекту при остеопетрозі з нирковим канальцевим ацидозом та церебральною кальцифікацією внаслідок дефіциту ізоензиму ІІ карбоксиангідрази–фермента, що каталізує синтез вугільної кислоти із вуглекислого газу та води. Вугільна кислота, дисоціюючи, утворює протони, які і створюють необхідне для нормального процесу резорбції у кісткових лакунах кисле середовище. Зниження активності фермента, таким чином, порушуватиме процес кісткової резорбції.
Основні діагностичні критерії:
Залежно від клінічних особливостей та часу маніфестації вирізняють інфантильну, проміжну та дорослу форми захворювання. Описано також цілий ряд більш рідкісних форм (летальна, транзиторна та ін.).
Діагностика:
Інфантильний остеопетроз злоякісний за перебігом, діагностується у ранньому дитинстві. Характерними ознаками є:
- затримка фізичного розвитку;
- дефекти кісток: підвищена ламкість та схильність до переломів, genu valgum, мандибулярний остеомієліт, деформація параназального синуса, сосковидного відростка, ураження лицьових нервів за рахунок защемлення їх у природних отворах черепа, специфічні лицьові прояви – гідроцефалія, проптоз, глухота;
- ураження кісткового мозку за рахунок заміщення його кістковою тканиною приводить до панцитопенії, що клінічно проявлятиметься анемією, кровотечами (тромбоцитопенія), схильністю до повторних інфекцій (лейкопенія), гепатоспленомегалією, гіперспленізмом та гемолізом (екстрамедулярний гемопоез);
- нічні апноє, сліпота внаслідок дегенерації сітківки.
Остеопетроз дорослого типу, або так званий доброякісний, діагностується у старшому підлітковому або дорослому віці. Майже у половини пацієнтів він має безсимптомний перебіг і виявляється випадково (при рентгенологічному обстеженні, або з огляду на обтяжений сімейний анамнез). У інших дебютує остеомієлітом (у 10% зустрічається мандибулярний остеомієліт) чи повторними переломами (близько 40% пацієнтів), болями в кістках, остеоартритами, карпальним тунельним синдромом. Кістковий мозок не скомпроментований.
Враховуючи ряд рентгенологічних, біохімічних та клінічних особливостей, виділяють дві форми остеопетрозу дорослого типу – тип І та тип ІІ.
Параклінічні обстеження:
- Лабораторні обстеження
- Інфантильний остеопетроз:
- Рівень сироваткового кальцію загалом відображає рівень його споживання із їжею. Виражена гіпокальцемія та рахітичні прояви виникають при значному дефіциті кальцію у раціоні.
- Часто характерний вторинний гіперпаратиреоз (підвищення рівня паратгормону).
- Підвищення рівня кислої фосфатази.
- Підвищення рівня креатинкінази-ВВ.
- Дорослий тип остеопетрозу:
- Рівень креатинкінази-ВВ та кислої фосфатази часто підвищений при остеопетрозі ІІ типу.
- Сироваткова (кістково-специфічна) лужна фосфтаза також може зростати.
- Інфантильний остеопетроз:
- Рентгенологічна симптоматика:
- генералізований остеосклероз;
- потовщення та ущільнення кісток черепа;
- зменшення розмірів та пневматизації синусів;
- виражене підвищення щільності хребців;
- переломи та остеомієліт.
МТР використовується для контролю лікування після трансплантації кісткового мозку.
Оскільки рентгенологічні прояви захворювання достатньо патогномонічні, біопсія кісток не обов’язкова.
Гістологічне дослідження корисне при відборі пацієнтів для трансплантації кісткового мозку.
Популяційна частота:
Близько 1 на 100000-500000, проте реальна частота невідома через відсутність епідеміологічних досліджень.
Співвідношення між статтю:
Остеопетроз зустрічається з однаковою частотою у осіб обох статей.
Генетичний ризик:
Остеопетроз дорослого віку І та ІІ типу (ОMIM 166600, 607634) успадковується за аутосомно-домінантним типом успадкування із відповідно 50% ризиком для потомства уражених пацієнтів. Усі інші типи остеопетрозу успадковуються за аутосомно-рецесивним типом з високим (25%) рекуррентним ризиком для сібсів.
Диференційна діагностика:
- пікнодизостоз;
- остеобластні метастази;
- отруєння фторидами;
- отруєння берилієм;
- лейкоз;
- серповидно-клітинна анемія.
Лікування та нагляд:
Остеопетроз інфантильного типу:
- Кальцитріол у великих дозах на фоні обмеженого вживання кальцію часом має виражений терапевтичний ефект за рахунок стимуляції остеокластів. Проте у більшості випадків він демонструє переважно клінічне покращення, котре не зберігається при припиненні лікування.
- Для корекції анемії застосовується еритропоетин.
- Глюкокортикоїди – тривало (1-2 мг/кг/день преднізолону) з метою стимуляції кісткової резорбції та лікування анемії.
- Гамма-інтерферон покращує функцію лейкоцитів, запобігає рецидиву інфекції.
- Хірургічне лікування передбачає трансплантацію кісткового мозку (при інфантильному типі), естетичну корекцію при виражених лицьових деформаціях, при переломах.
Специфічних методів лікування дорослого типу остеопетрозу не розроблено. Останній вимагає медичного втручання переважно при розвитку ускладнень.
Пренатальна діагностика: специфічна не розроблена.
Прогноз:
Прогноз у випадку нелікованого інфантильного остеопетрозу стосовно життя серйозний. Пацієнти гинуть протягом перших 10 років життя від вираженої анемії, кровотеч чи інфекцій. Прогноз при остеопетрозі дорослого типу сприятливий.
Профілактика:
Медико-генетичне консультування у сім’ях з ускладненим щодо остеопетрозу анамнезом, генетичне тестування, а також профілактичні заходи для попередження можливих ускладнень.
Номер з каталогу МІМ:
- 259700 Osteopetrosis, Autosomal Recessive 1; OPTB1.
- 166600 Osteopetrosis, Autosomal Dominant 2; OPTA2.
- 259730 Osteopetrosis Autosomal Recessive 3; OPTB3.
- 607634 Osteopetrosis, Autosomal Dominant 1; OPTA1.
- 600329 Osteopetrosis and Infantile Neuroaxonal Dystrophy.
- 259710 Osteopetrosis Autosomal Recessive 2; OPTB2.
- 259720 Osteopetrosis Autosomal Recessive 5; OPTB5.
Література:
- Albers-Schonberg H: Roentgenbilder einer seltenen Knochennerkrankung. Munch Med Wochenschr 1904;51:365.
- Armstrong DG, Newfield JT, Gillespie R. Orthopedic management of osteopetrosis: results of a survey and review of the literature. J Pediatr Orthop 1999;19(1):122-32.
- Beighton P, Hamersma H, Cremin BJ. Osteopetrosis in South Africa. The benign, lethal and intermediate forms. S Afr Med J 1979;55(17):659-65.
- El-Tawil T, Stoker DJ. Benign osteopetrosis: a review of 42 cases showing two different patterns. Skeletal Radiol 1993;22(8):587-93.
- Felix R, Hofstetter W, Cecchini MG: Recent developments in the understanding of the pathophysiology of osteopetrosis. Eur J Endocrinol 1996;134(2):143-56.
- Jones K.L. Smith’s recognizable patterns of human malformation. W.B. Saunders company 1988:372–374.
- Key L, Carnes D, Cole S. Treatment of congenital osteopetrosis with high-dose calcitriol. N Engl J Med 1984;310(7):409-15.
- Manusov EG, Douville DR, Page LV. Osteopetrosis (‘marble bone’ disease). Am Fam Physician 1993;47(1):175-80.
- Warkany J. Congenital Malformations, Notes and Comments. Year book medical publishers 1971:285-289.
- Whyte MP. Sclerosing Bone Dysplasias. In: Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. 1999:367-383.
Переглянуто редакційною колегією I.B.I.S.: 27/08/2003
Дивіться також:
- Остеопетроз (інформація для батьків)
Довжина довгих трубчастих кісток у хлопчиків (продовження)
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”
Чоловіча стать – коливання розмірів трубчастих кісток за даними рентгенографії (в см)
Кістки ноги
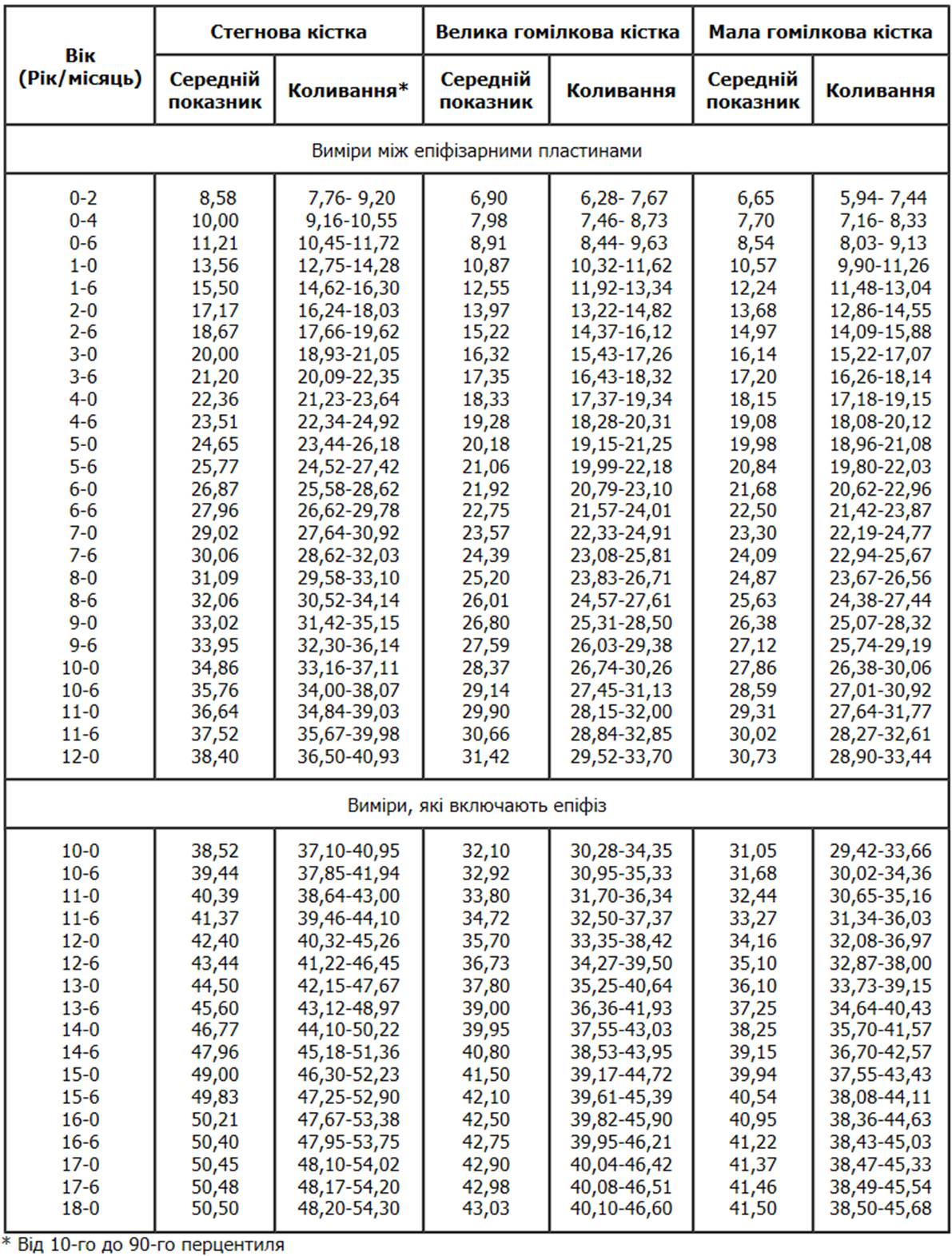
Радіографічні показники, отримані в результаті обстежень 175 осіб віком від 2 місяців до 18 років, які проживали в м. Денвер, штат Колорадо. Усі обстежені були білої раси, за своїм соціальним статусом відносились до середнього класу, походженням переважно з північної Європи. Maresh MM: Am J Dis Child 89:725, 1955.
Довжина довгих трубчастих кісток у хлопчиків
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”
Чоловіча стать – коливання розмірів трубчастих кісток за даними рентгенографії (в см)
Кістки руки
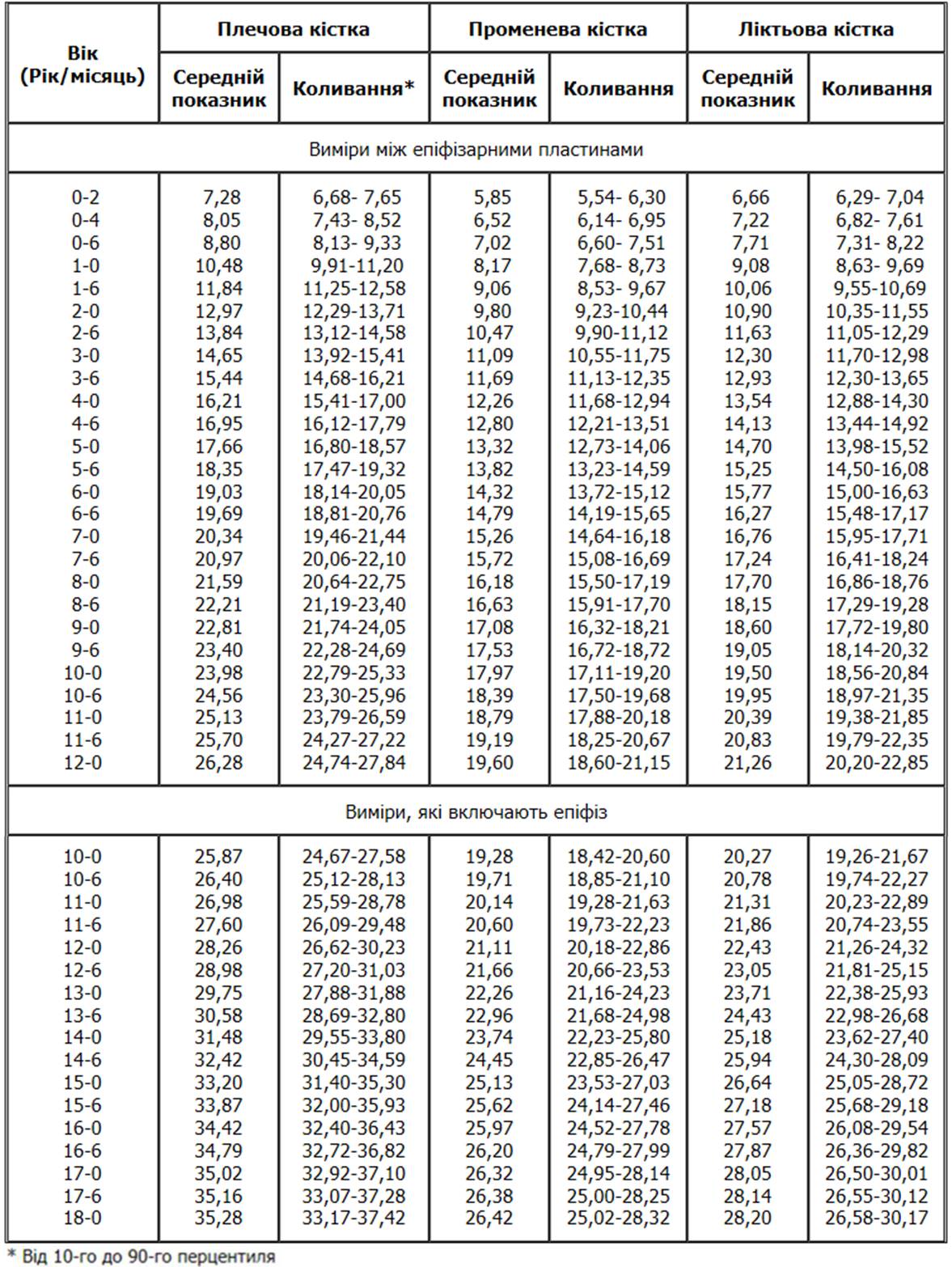
Радіографічні показники, отримані в результаті обстежень 175 осіб віком від 2 місяців до 18 років, які проживали в м. Денвер, штат Колорадо. Усі обстежені були білої раси, за своїм соціальним статусом відносились до середнього класу, походженням переважно з північної Європи. Maresh MM: Am J Dis Child 89:725, 1955.
Омфалоцеле
(Omphalocele)
П.О. Галайко
Студент Буковинської
державної медичної академії
Визначення:
Омфалоцеле або вроджена пупкова кила – це дефект передньої стінки черевної порожнини в ділянці пупкового кільця, з утворенням килового мішка, вкритого амніоперитонеальною мембраною, в якому містяться органи черевної порожнини.
Популяційна частота: 1:4000 – 1:6000.
В ізольованій формі омфалоцеле спостерігається в 46% випадків, але крім цього воно входить до складу таких вроджених аномалій, як пентада Кантрелла (серединний надпупковий дефект черевної стінки, дефект нижньої частини грудини, недостатність діафрагмальної частини перикарду, недостатність передньої частини діафрагми, серцеві аномалії), синдром Беквіта-Відеманна (макроглосія, омфалоцеле і вісцеромегалія); як правило, омфалоцеле виникає у комбінації з хромосомними абераціями (35-58% випадків), зокрема з трисоміями 13, 18 і 21, синдромом Тернера (45,Х), триплоїдіями. Можливі випадки успадкування омфалоцеле по аутосомно-домінантному, рецесивному, зчепленому зі статтю і полігенному типах, залежно від приєднання інших симптомів. Також омфалоцеле може комбінуватися з аномаліями серцево-судинної системи (дефекти міжшлуночкової та міжпередсерної перегородок, тетрада Фалло) у 47% випадків, аномаліями сечостатевої системи у 40% випадків, дефектами нервової трубки у 39% випадків. У 20% випадків омфалоцеле спостерігається внутрішньоутробна затримка розвитку плода.
Співвідношення статей: Ж1 : Ч1.
Патогенез:
В основі виникнення вади лежить порушення в ембріогенезі формування черевної стінки внаслідок недостатності однієї з чотирьох ектомезодермальних закладок (головна, каудальна, дві латеральні). Таким чином недостатність латеральних закладок, що в нормі з’єднуються по серединній лінії між 3-м і 4-м тижнями, призводить до виникнення ізольованого омфалоцеле. Недостатність головної закладки при злитті з іншими закладками призводить до виникнення омфалоцеле з ектопією серця, дефектами грудини і діафрагми. Недостатність каудальної закладки призводить до екстрофії сечового міхура.
Типи омфалоцеле:
- епігастральне (дефект головної закладки);
- класичне (дефект латеральної закладки);
- гіпогастральне (дефект каудальної закладки).
Поєднані аномалії:
- Тетрада Фалло (33%);
- вторинний дефект міжпередсердної перетинки (19%);
- дефект міжшлуночкової перетинки (ДМШП), каорктація аорти, спільний артеріальний стовбур, атріо-вентрикулярна комунікація (зрідка).
Генетичні синдроми:
- трисомія 13, 18, 21 хромосом;
- синдром Беквіта-Відемана (ОМІМ 130650);
- Шпрінцена синдром (ОМІМ 182210);
- Летальний синдром омфалоцеле – щілини піднебіння (ОМІМ 258320);
- OEIS (омфалоцеле, екстрофія, неперфорований анус, спінальні дефекти) – ОМІМ 258040.
Основні клінічні ознаки:
- Класичне омфалоцеле: дефект локалізується по серединній лінії, а вміст черевної порожнини випирає через пупкове кільце. Вміст кили представлений петлями кишківника, шлунком і печінкою, які вкриті мембраною, що складається з двох шарів: внутрішнього (очеревина) і зовнішнього (амніон). До складу килового мішка входить також пуповина. Розміри дефекту варіюють від незначних, що містять кілька петель кишківника, до великих, у просвіт яких потрапляє більшість органів черевної порожнини.
- Епігастральне омфалоцеле (пентада Кантрелла):
- розчеплення грудини;
- вроджена вада серця (ВВС) – аномалія перикарду, ектопія серця;
- дефекти діафрагми серединні;
- епігастральне омфалоцеле.
- Гіпогастральне омфалоцеле:
- агенезія товстого кишківника;
- екстрофія сечового міхура;
- неперфорований анус;
- везико-інтестинальна нориця.
Ускладнення:
- затримка внутрішньоутробного розвитку (20%);
- недоношеність (10-50%);
- малюкова смертність (47-60%);
- інші поєднані аномалії (30-70%);
- ВВС (20-40%).
Пренатальна діагностика:
- УЗД-виявлення об’ємних утворень, що прилягають до передньої черевної стінки плода і містять внутрішні органи.
- Скринінг сироваткових маркерів: дефекти передньої черевної стінки можуть призводити до підвищення рівня альфа-фетопротеїну (АФП) у сироватці крові вагітної. Встановлено, що скринінг АФП у 52% випадків сприяє виявленню дефектів черевної стінки і у 42% випадків – виявленню омфалоцеле.
Нижче приведені фотографії двох випадків омфалоцеле, виявлених при УЗД в ІІ триместрі вагітності.
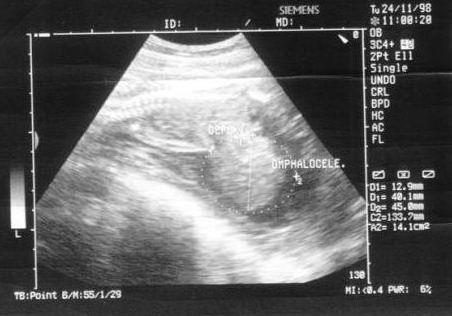
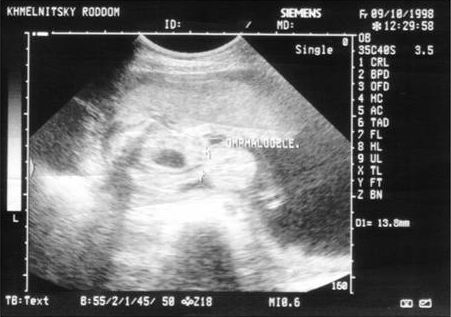
Диференційний діагноз:
Насамперед слід проводити із гастрошизисом. Омфалоцеле локалізується по серединній лінії, пуповина входить в киловий мішок, органи вкриті мембраною, яка зв’язана з пуповиною. До винятків відносяться випадки омфалоцеле, при яких відбувається внутрішньоутробний розрив амніоперитонеального мішка, але таке ускладнення зустрічається досить рідко.
Нагляд та опіка:
- Первинна реанімація за схемою АВС:
- інтубація;
- вентиляція та оксигенація;
- відновлення рідинного балансу.
- Температурний контроль:
- уникати значної тепловтрати.
- Запобігання інфікування:
- антибіотики (ампіцилін та гентаміцин).
- Чітко віддиференціювати омфалоцеле від гастрошизису:
- дбайливо помістити киловий мішок, уникати травмування;
- підтримувати без натягу;
- декомпресія шлунково-кишкового тракту (ШКТ) через назо-гастральний зонд;
- ультразвукове обстеження (УЗО) для уточнення походження органів килового мішка.
- Пошук асоційованих вад:
- пентада Кантрелла;
- ВВС;
- трисомії хромосом;
- синдром Беквіта-Відемана.
Лікування:
Невеликий дефект черевної стінки може бути закритий за допомогою одноетапної операції. Великі дефекти усуваються, як правило, при проведенні двохетапної операції (Schuster, 1967), з використанням силіконової чи тефлонової мембрани для закриття отвору і усунення евентрації внутрішніх органів.
Нехірургічні методи лікування (Grob, 1957) показані у випадках множинних вроджених вад розвитку (МВВР), що не підлягають корекції і передбачають обробку омфалоцеле нітратом срібла, 65% спирту. Однак летальність у цих випадках становить понад 33%, епітелізація триває 8-10 тижнів.
Прогноз:
Прогноз при омфалоцеле, в основному, залежить від наявності супутніх аномалій. Перинатальні втрати пов’язані з серцевими вадами, хромосомними порушеннями і недоношеністю.
При виявленні омфалоцеле у плода потрібно проводити ретельний пошук супутніх аномалій. Показане визначення каріотипу і проведення ехокардіографічного обстеження. При виявленні вади до настання періоду життєздатності плода можна рекомендувати переривання вагітності. При прогресуючій вагітності і виявленні омфалоцеле в ІІІ триместрі показане ультразвукове дослідження в динаміці для визначення ознак кишкової непрохідності і внутрішньоутробної затримки розвитку плода. Суперечливим сьогодні залишається питання про оптимальний метод родорозрішення при омфалоцеле плода. Деякі спеціалісти вважають, що родорозрішення в таких випадках повинно здійснюватись за допомогою операції кесарського розтину для запобігання розриву амніоперитонеального мішка. Однак, за даними ретроспективного аналізу, результат після кесарського розтину не відрізнявся від результату родорозрішення через природні пологові шляхи.
Запобігання:
- медико-генетичне консультування;
- пренатальна діагностика.
Номер з каталогу МІМ:
Література:
- Baird, P. A.; MacDonald, E. C. : An epidemiologic study of congenital malformations of the anterior abdominal wall in more than half a million consecutive live births. Am. J. Hum. Genet. 33: 470-478, 1981.
- Calzolari, E.; Bianchi, F.; Dolk, H.; Milan, M.; EUROCAT Working Group : Omphalocele and gastroschisis in Europe: a survey of 3 million births 1980-1990. Am. J. Med. Genet. 58: 187-194, 1995.
- Hershey, D. W.; Haesslein, H. C.; Marr, C. C.; Adkins, J. C. : Familial abdominal wall defects. Am. J. Med. Genet. 34: 174-176, 1989.
- Kallen, K.; Castilla, E. E.; Robert, E.; Mastroiacovo, P.; Kallen, B. : OEIS complex–a population study. Am. J. Med. Genet. 92: 62-68, 2000.
- Lee, D. H.; Cottrell, J. R.; Sanders, R.C.; Meyers, C.M.; Wulfsberg, E.A.; Sun, C.-C.J.: OEIS complex (omphalocele-exstrophy-imperforate anus-spinal defects) in monozygotic twins. Am. J. Med. Genet. 84: 29-33, 1999.
- Nyberg, D.A., Mahony, B.S., and Pretorius, D.H. Abdominal Wall Defects in Diagnostic Ultrasound of Fetal Anomalies. Mosby-Year Book, Inc: St Louis, 1990: 395-432.
- Nyberg, D.A., Mahony, B.S., and Pretorius, D.H. Intra-Abdominal Abnormalities in Diagnostic Ultrasound of Fetal Anomalies. Mosby-Year Book, Inc: St Louis, 1990: 342-394.
- Romero, R., Pilu, G., Jeanty, P., Ghidini, A., and Hobbins, J.C. The Abdominal Wall in Prenatal Diagnosis of Congenital Abnormalities. Appleton & Lange: Connecticut, 1988: 209-232.
- Romero, R., Pilu, G., Jeanty, P., Ghidini, A., and Hobbins, J.C. The Gastrointestinal Tract and Intraabdominal Organs in Prenatal Diagnosis of Congenital Abnormalities. Appleton & Lange: Connecticut, 1988: 233-254.
- Smith, N. M.; Chambers, H. M.; Furness, M. E.; Haan, E. A. : The OEIS complex (omphalocele-exstrophy-imperforate anus-spinal defects): recurrence in sibs. J. Med. Genet. 29: 730-732, 1992.
- Yang, P.; Beaty, T. H.; Khoury, M. J.; Chee, E.; Stewart, W.; Gordis, L. : Genetic-epidemiologic study of omphalocele and gastroschisis: evidence for heterogeneity. Am. J. Med. Genet. 44: 668-675, 1992.
Переглянуто редакційною колегією I.B.I.S.: 24/12/2002
Хвороба Норрі
(Norrie Disease)
Наталія Василівна Гузюк
Офтальмолог
Волинського обласного дитячого
територіального медичного об’єднання
Синоніми:
Вроджена двобічна псевдогліома сітківки; Atrophia Bulborum Hereditaria, Episkopi Blindness. Включає: X-linked Familial Exudative Vitreoretinopathy (X-Linked FEVR).
Мінімальні діагностичні ознаки: двобічна дисплазія сітківки.
Клінічна характеристика:
Характеризується спектром фіброзних і васкулярних змін сітківки, що прогресують до підліткового віку і призводять до зорових порушень різного ступеня. Найбільш важкими змінами сітківки є сіро-жовті фіброваскулярні маси, які з’являються в перші декілька місяців життя, результатом чого є повна сліпота. Менш складними порушеннями є фіброзний білий тяж, що відходить від диску зорового нерва і прикріплюється до кришталика (первинне персистуюче гіперпластичне скловидне тіло) і периферичні ретинальні васкулярні аномалії з чи без фіброзних змін (сімейна ексудативна вітреоретинопатія).
Діагностика:
Хвороба Норрі діагностується на основі характерних ретинальних змін в осіб чоловічої статі, сімейної історії і молекулярно-генетичних досліджень гена NDP (локус хромосоми Хр.11.4), що виявляють мутації, які призводять до хвороби, приблизно у 85% уражених чоловіків. Більшість мутацій є унікальними точковими мутаціями, але приблизно в 15% пацієнтів було виявлено інтратрігенні і субмікроскопічні делеції, включаючи NDP.
Генетична консультація:
Тип успадкування Х-зчеплений рецесивний. Жінку-носія визначають через дослідження родоводу і аналіз мутацій гена NDP, в тому випадку, коли мутацію, що призводить до хвороби було виявлено в уражених родичів чоловічої статі. Хворий батько передає мутацію своїм дочкам, але ні одному із синів. Передача мутації жінкою-носієм кожній дитині складає 50%. У чоловіків така мутація спричиняє хровобу Норрі, а жінки є носіями. Пренатальна діагностика є можлива у тому випадку, коли мутація, що викликає хворобу була виявлена у жінки-носія.
Гетерозиготи:
В деяких випадках жінки-носії можуть мати деякі ретинальні порушення (відшарування сітківки, аномальну васкуляризацію сітківки з втратою зору). Деякі жінки-носії можуть мати легку сенсоневральну втрату слуху.
Клінічна діагностика:
Мутації в гені NDP є асоційованими зі спектром ретинальних змін в межах від хвороби Норрі до Х-зчепленої сімейної ексудативної вітреоретинопатії, включаючи деякі випадки первинного персистуючого гіперпластичного скловидного тіла, хвороби Коутса і прогресуючої ретинопатії недоношених. Зрідка у жінок-носіїв можуть бути зміни сітківки.
Очні зміни, які виявляються при хворобі Норрі:
- Білатеральні, часто симетричні, ураження очей.
- Очі нормального розміру, з нормальними передніми камерами і чистими кришталиками при народженні.
- Аномалії скловидного тіла (крововиливи, мембрани, відшарування, вітреоретинальні маси).
Табл.1. Класифікація фенотипів ока, виявлених при хворобі Норрі.
Вік |
Вік |
||
|---|---|---|---|
З народження до 3 міс. |
Від 3 міс. до 8-10 років. |
||
З народження. |
|||
З народження. |
До 20 років. |
||
З народження. |
Клінічний опис:
Очні зміни: у чоловіків з хворобою Норрі є зазвичай білатеральними і симетричними. Вони є присутніми при народженні і як правило прогресують. З відкриттям NDP-гену стало відомо, що фенотиповий спектр очних аномалій хвороби Норрі є широким (табл.1). Фенотип коливається від класичної хвороби Норрі до сімейної ексудативної вітреоретинопатії (СЕВ), включаючи деякі випадки первинного персистуючого гіперпластичного скловидного тіла (ППГСТ) і важкої ретинопатії недоношених (РН).
Класичний очний фенотип хвороби Норрі: зміни на очному дні проявляються з народження – в скловидному тілі і сітківці формуються сіро-жовті блискучі фіброваскулярні маси, субретинальна ексудація призводить до відшарування сітківки, в ній з’являються крововиливи; до кінця 2 місяця життя розвивається тотальне неоперабельне відшарування сітківки. При народженні рогівка, передня камера ока, райдужна оболонка, внутрішньоочний тиск і розмір очного яблука в межах норми. З 3 місяців до 8-10 років, як правило, виникають прогресуючі зміни, які проявляються помутнінням кришталика (катарактою), атрофічними змінами райдужної оболонки з передніми і задніми сенехіями, передня камера стає мілкою, виникає оклюзія шляхів відтоку внутрішньоочної рідини, результатом чого є підвищення внутрішньоочного тиску. За цими змінами виникає помутніння рогівки та стрічкоподібна кератопатія, втрата внутрішньоочного тиску, результатом чого є зменшення розмірів очного яблука (phthisis bulbi). На кінцевій стадії хвороби Норрі рогівки стають молочного кольору, а очі часто западають в орбіти.

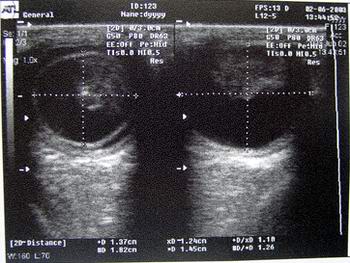
Зовнішній вигляд дитини з хворобою Норрі і результат УЗ-обстеження.
Праве очне яблуко – 1,37 х 1,24 см. Ліве очне яблуко – 1,82 х 1,45 см.
В передньо-медіальних відділах очних яблук візуалізуються підвищеної
ехогенності, неправильної форми, гетерогенні включення
- Когнітивні зміни (зміни поведінки): біля 50% осіб чоловічої статі з фенотипом хвороби Норрі є розумово відсталими з характерними змінами поведінки. Когнітивні зміни є варіабельними як внутрісімейно, так і міжсімейно.
- Слухові зміни: слуховий нерв уражається в 40% випадків (в основному особи чоловічої статі), при цьому розвивається нейросенсорна приглухуватість. Втрата слуху відбувається в ранньому дитинстві, вона може бути поступово прогресуючою або раптовою. За аудіологічними даними патологічний процес локалізується в завитку (внутрішнє вухо), тоді як позазавиткові функції і функції коркового центру збережені. Втрата слуху не завжди супроводжується когнітивними змінами.
- Інше: змін з боку інших органів немає, період життя може бути скороченим за рахунок загального ризику пов’язаного з розумовою відсталістю, сліпотою і/чи втратою слуху.
ППГСТ характеризується фіброзним білим тяжем з судинами, який відходить від диска зорового нерву і прикріплюється до задньої капсули кришталика з темпоральної сторони. Сітківка може бути в складках, або відшарована; кришталик може бути прозорим або ні.
СЕВ характеризується ранньою зупинкою васкуляризації сітківки, результатом чого є наявність аваскулярних зон на периферії сітківки. Ретинальні зміни можуть бути пов’язані як з наявністю аваскулярних зон на периферії сітківки, так і вродженими серпоподібними ретинальними складками чи відшаруванням сітківки, зумовленими субретинальною проліферацією і ексудацією. Якщо дані зміни знаходяться в центральних відділах очного дна – вони призводять до тракційної деформації диска зорового нерва та скроневої гетеротопії макули.
Молекулярно-генетичні дослідження:
Ген NDP є єдиним відомим на даний час геном, пов’язаним із хворобою Норрі. На даний час відомо більше ніж 85 місенс, нульових і сплайс мутацій, а також субмікроскопічні та інтрагенні делеції. Більшість цих мутацій є унікальні і виявляються в поодиноких пацієнтів. Біля 15% мутацій є субмікроскопічні делеції. Основні мутації не є індифіковані. Методи: пряма діагностика мутацій.
- Сіквенс-аналізи. Дозволяє достовірно виявити часткові чи повні делеції гена. Приблизно 85% пацієнтів, які мають мутації гена NDP, виявлять за цим методом.
- Блот-гібридизація по Саузерну. Можлива блот-гібридизація по Саузерну в наукових установах, яка дозволяє виявити субмікроскопічні делеції в чоловіків і носіїв-жінок.
Гістопатологія:
В сітківці виявляють недорозвинуті ретинальні клітини, які спричинені ембріональним недорозвитком сітківки. Ретинальна васкулярна система є також недорозвинутою.
Лікування:
З народження у більшості чоловіків з класичним фенотипом хвороби Норрі відмічається тотальне відшарування сітківки. Інтервенційна терапія мало що може дати щодо збереження зору. При прогресуванні хвороби Норрі, яке проявляється підвищенням очного тиску, виникає потреба у хірургічному втручанні. Зрідка необхідна енуклеація ока із-за виражених больових відчуттів. Якщо діагностоване часткове відшарування сітківки, то хірургічне втручання чи лазерна терапія можуть бути помічними. Рутинне офтальмологічне обстеження необхідно проводити усім хворим хворобою Норрі, навіть коли гострота зору серйозно знизилась. Більшість хворих з хворобою Норрі є сліпими і потребують обов’язкового обстеження слуху з ціллю ранньої корекції глухоти. Слуховий апарат, зазвичай, є помічним у зрілому віці і в старості. Кохлеарні імплантанти слід брати до уваги, коли значно порушений слух. Питання поведінки є проблемним на все життя для пацієнтів з хворобою Норрі і їх опікунів, незважаючи на те, чи є розумова відсталість, чи присутні когнітивні порушення.
Медикаментозна терапія є симптоматичною.
Номер з каталогу МІМ:
310600 Norrie Disease; ND.
Література:
- Katherine B Sims, MD. NDP-Related Retinopathies (U.S. National Library of Medicine).
- Norrie Disease; ND. OMIM #310600.
Переглянуто редакційною колегією I.B.I.S.: 23/12/2004
Довжина ступні
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”

Дані отримані в результаті обстеження 227 дівчат та 285 хлопчиків віком від 1 до 18 років, які проживали в м. Бостоні, штат Массачусетс. Всього було проведено 3128 вимірювань. Усі ці діти перенесли поліомієліт і мали ураження лише однієї нижньої кінцівки. Вимірювались лише здорові кінцівки. Лінійний повздовжний ріст ноги зазвичай передує підлітковому росту і закінчується за два-три роки до досягнення остаточного дорослого зросту. Blais MM et al: J Bone Joint Surg 38A:998, 1956.

Вимірювання руки (продовження)
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”



Дані базуються на популяційному дослідженні 2403 дітей від народження до 14 років. 56% становили діти чоловічої статі, 44% – жіночої; 2006 відносились до білої раси, 206 – до темношкірої, 43 – азійської. Feingold M, Bossert WH: Birth Defects: Orig Art Series X(13), 1974.
|
|
|
|
|
|




