
Serhiy
Преконцепційна профілактика
(Preconception Prevention)
Олександр Романович Колтонюк
лікар акушер-гінеколог, лікар УЗД
Рівненського обласного клінічного лікувально-діагностичного центру ім. В. Поліщука
Висока частота вроджених вад розвитку (ВВР), самовільних викиднів та передчасних пологів являє серйозну медичну проблему, а їхнє попередження й рання діагностика є винятково актуальними. У структурі перинатальної смертності частка вроджених аномалій розвитку становить близько 25%. В основі багатьох спадкових захворювань лежать хромосомні й генні мутації, що виникають у статевих клітинах у процесі гаметогенезу, при заплідненні й на перших стадіях дроблення зиготи. Більшість аномальних ембріонів елімінуються в перші дні після зачаття. Однак збої в механізмах так званого “природнього добору” визначають збереження й розвиток патологічної вагітності й народження хворого потомства. Зниження частоти патологічних зачать має істотне значення для профілактики вродженої патології й репродуктивних втрат. Основна мета преконцепційної профілактики полягає в створенні оптимальних умов для дозрівання гамет, овуляції, запліднення, імплантації й сприятливого розвитку ембріона.
Преконцепційна профілактика – це комплекс заходів, що проводяться за деякий час до зачаття, спрямованих на зменшення частоти вроджених вад розвитку (ВВР) та ускладненного перебігу вагітності (мимовільні викидні, передчасні пологи, народження дітей з вагою менше 2500 г. та ін.).
Покази до преконцепційної профілактики:
- Вік подружжя молодше 18 років і більше 35 років;
- Непліддя в анамнезі;
- Звичне невиношування й мертвонародження в анамнезі;
- Генетичний ризик мультифакторіальних вад розвитку;
- Народження гіпотрофічних дітей і передчасні пологи в анамнезі;
- Інфекційні захворювання (краснуха, токсоплазмоз, цитомегаловірус, герпес та інші вірусні інфекції);
- Цукровий діабет, фенілкетонурія та інші ендокринні й обмінні захворювання;
- Екстрагенітальні захворювання (гіпертонічна хвороба, захворювання нирок, кардіопатія тощо);
- Тривале вживання лікарських препаратів (у тому числі оральних контрацептивів, цитостатиків, антидепресантів, іммунодепресантів, протисудомних та інших препаратів).
Діагностичні заходи в рамках преконцепційної профілактики проводяться на етапі підготовки до планованої вагітності. Учасниками преконцепційної діагностики є: гінеколог, генетик, терапевт, андролог, ендокринолог.
Преконцепційна діагностика включає в себе:
- Консультація лікаря–генетика, генетичні дослідження (анамнез, збір родоводу, дослідження каріотипу, ДНК-діагностика тощо).
- Гінекологічне дослідження:
- Анамнез і вагінальне дослідження;
- Обстеження на запальні захворювання органів малого тазу;
- Гормональні дослідження й вимірювання базальної температури з метою уточнення характеру порушень менструального циклу (ановуляція, недостатність лютеїновой фази, гіпоестрогенія, гіперпролактинемія);
- Визначення рівня антифосфоліпідних і антиспермальних антитіл.
- Обстеження на інфекції:
- Обстеження на TORCH-інфекції (токсоплазмоз, краснуха, цитомегаловірус, герпес та ін.). Покази до обстеження на інфекції групи TORCH у преконцепційному періоді:
- первинне і вторинне непліддя;
- самовільні викидні в анамнезі;
- мертвонародження і “завмерлі” вагітності;
- аномалії розвитку плода в анамнезі;
- багатоводдя, маловоддя, ураження плаценти, УЗ-маркери внутрішньоутробного інфікування при попередніх вагітностях;
- наявність ознак інфекційного ураження у новонародженого в анамнезі;
- клінічні прояви TORCH–інфекції у жінки (запальні хвороби сечостатевої системи, артрит, кон’юнктивіт, тощо);
- належність до групи професійного ризику (медичні працівники, робітники м’ясопереробної промисловості та ін.).
- Обстеження на хламідіоз, уреаплазмоз, мікоплазмоз, папілломавіруси, гарднерели, кандидоз, стрептококки группи В, золотистий стафілококк (staph. aureus), ентерококки, кишечну паличку.
- Обстеження на TORCH-інфекції (токсоплазмоз, краснуха, цитомегаловірус, герпес та ін.). Покази до обстеження на інфекції групи TORCH у преконцепційному періоді:
- Терапевтичне обстеження.
- Ендокринологічне обстеження (з метою виключення цукрового діабету, гіпо- та гіпертиреозу та ін.).
Метою преконцепційної профілактики є створення оптимальних умов для дозрівання яйцеклітини, її сприятливого розвитку та імплантації.
Заходи преконцепційної профілактики:
- Щоденний прийом фолієвої кислоти в таблетках у дозі 0,4 мг:
- Міністерство охорони здоров’я України рекомендує усім жінкам репродуктивного віку щоденно приймати фолієву кислоту (вітамін В9) в таблетках у дозі 0,4 мг.
- Жінкам групи ризику виникнення повторних випадків вад невральної трубки при плануванні вагітності рекомендовано приймати фолієву кислоту периконцепційно (за 3 місяці до зачаття і перші 2-3 місяці вагітності) у дозі 4 мг щодня!
- Раціональне харчування. Має значення споживання їжі багатої фолатами й вітамінами групи В. Харчові джерела фолієвої кислоти:
- Злакові й продукти їхньої переробки (борошно грубого помолу, хлібо-булочні виробу із цього борошна, гречана й вівсяна крупа);
- Квасоля, соя, цвітна капуста, салат, шпинат, зелена цибуля, гриби;
- Печінка, сир, ікра;
- Яйця.
- Прийом мультивітамінних комплексів, що містять B12, В6 С, Е щонайменше три місяці до зачаття й протягом першого триместру вагітності.
- Обмеження нічних, змінних і вахтових робіт, що супроводжуються зміною фотоперіодизації, і призводять до зсуву овуляції та внутріфолікулярного старіння яйцеклітин.
- Виключення побутових (паління, алкоголь, недоброякісних або сумнівних продуктів харчування) і промислових токсикантів (розчинники, летючі речовини та ін.), шкідливих фізичних факторів (шум, вібрація, електромагнітні поля, іонізуюча радіація та ін.). Відмова від гарячих ванн, саун, занять фізично важкими видами спорту.
- Виключення надмірних стресових подразників.
- Нормалізація менструального циклу й секреторних перетворень.
- Синхронізація репродуктивних процесів (планування зачаття на літньо-осінній період, вибір часу зачаття відповідно до овуляції), що дозволяє запобігти “старінню” статевих клітин.
- Повна відмова від вживання алкогольних напоїв, особливо під час вагітності, що дозволить на 100% запобігти розвитку алкогольного синдрому плода.
- Превентивне лікування виявленої патології з подальшим контролем його ефективності.
- Хворі на фенілкетонурію жінки перед плануванням вагітності повинні обов’язково проконсультуватись у лікаря-генетика для запобігання фенілаланіновій ембріофетопатії.
- Вакцинація від краснухи (червонички), що запобігає розвитку синдрому вродженої краснухи.
Внутрішньоутробний розвиток – один з найбільш відповідальних періодів у житті людини. Саме тому, раціонально використовуючи преконцепційну профілактику, можна досягти створення найкращих умов для розвитку плода, що дозволяє істотно знизити ризик вроджених вад і внутрішньоутробних інфекцій. Ретельно проведена преконцепційна профілактика дозволяє у 3(!) рази підвищити вірогідність настання нормальної вагітності, виношування та народження здорової дитини.
Переглянуто редакційною колегією I.B.I.S.: 19/05/2008
Обвід голови та грудної клітки у близнюків
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”
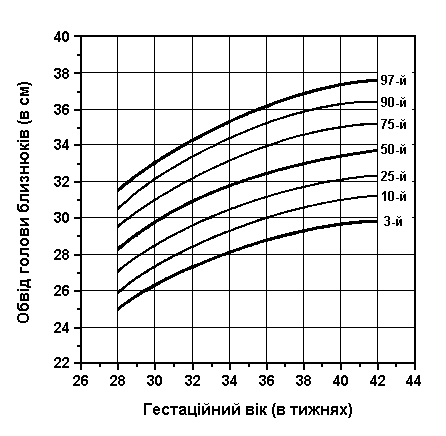
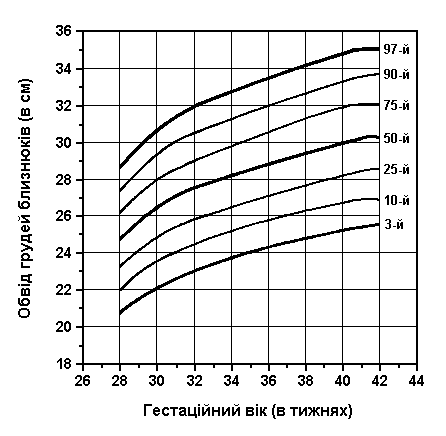
Обвід голови і грудей близнюків при народженні (3-й, 10-й, 25-й, 50-й, 75-й, 90-й та 97-й персентилі) був визначений Leroy et al (Acta Genet Med Gemellol 31:199, 1982). Це французське дослідження 1049 близнюків включало дані про тих близнюків, що народилися з гестаційним віком від 28 до 42 тижнів, та поєднувало дані без зазначення зиготності, статі та даних про народження.
Порфірія
(Porphyria)
Н.С. Шкапій
Спеціаліст з інформаційного забезпечення
Хмельницького ОМНІ-центру
Що таке порфірія?
Термін порфірія об’єднує щонайменше вісім окремих форм захворювання, що значно відрізняються одне від одного. Загальною ознакою для усіх порфірій є накопичення в організмі порфірину або його попередників. Ці речовини є нормальними продуктами обміну, однак при порфірії вони накопичуються у надмірній кількості.
Клінічні прояви різних типів порфірії не є однаковими. Форми лікування також залежать від прояву захворювання. Тому, дуже важко дати одну характеристику цій хворобі, яка б підходила для усіх її типів.
Оскільки симптоми порфірії неспецифічні, тобто характерні для багатьох інших захворювань, то діагностика її часто є запізнілою.
Спершу вражається нервова система та шкіра. Порушення роботи нервової системи є проявом гострої порфірії. Опіки, пухирі та рубці на ділянках шкіри, що найбільше піддаються дії сонячного проміння, є ознаками хвороби.
Терміни “порфірія” та “порфірин” походять від грецького “porphyrys”, що означає “фіолетовий”. Сеча деякий хворих на порфірію може мати червоний відтінок внаслідок надлишку в ній порфірину та пов’язаних із ним сполук. Під дією сонячного світла така сеча через деякий час темніє.
Як успадковується порфірія?
Кожний тип порфірії характеризується дефіцитом певного ензиму (фермента). Ці ферменти залучені у синтез гему – речовини, яка у великій кількості знаходиться у кістковому мозку, червоних кров’яних клітинах (еритроцитах) та печінці. Гем функціонує як гемоглобін у кістковому мозку та червоних кров’яних клітинах. Особи із аутосомно–домінантною формою порфірії (успадковуючи ген від одного з батьків) мають один “пошкоджений” ген, що поєднується із здоровим. “Аутосомні” гени завжди парні – по одному від кожного з батьків. Тому, в середньому, половина нащадків успадкує цей “пошкоджений” ген, а інша половина – “здоровий”. У декого із успадкованим геном хвороби з’являться симптоми захворювання. Пацієнти із аутосомно-рецесивним типом порфірії мають пару пошкоджених генів, а кожна їх дитина отримує один “пошкоджений” ген. Цей ген буде в парі із нормальними геном, тому ніякі симптоми захворювання не спостерігатимуться. Якщо у пари, що є носіями однакового аутосомно–рецесивного гену народжуватимуться діти, то приблизно 1 із 4 їхніх дітей успадкує 2 “хворих” гени і буде мати симптоми порфірії. Оскільки усі типи порфірії є відносно рідкісними, дуже мала вірогідність того, що більше ніж одна форма порфірії зустрінеться в одній сім’ї, чи один тип порфірії розвинеться в інший.
Як класифікується порфірія?
Найлогічнішим підходом до класифікації порфірії є класифікація за типом “пошкодженого” гена. Ферменти, яких не вистачає при порфірії зазвичай приймають участь у формуванні гему із простих молекул. Гем – це життєво необхідна речовина людського тіла, яка складається із атому заліза та молекули порфірину, що його оточує.
Іноді використовують і інші класифікації. Наприклад, часто розрізняють порфірії двох типів – печінкові та еритропоетичні. Гостра порфірія характеризується різким болем та іншими проявами, що пов’язані із нервовою системою. Такі ознаки хвороби можуть виникати раптово, або мати тривалий характер. Якщо в пацієнта виявлений дефіцит певного ферменту, але ніякі симптоми хвороби не спостерігаються, то такий тип порфірії називають латентною. Між латентною та гострою порфірією може бути широкий спектр прояву хвороби залежно від її типу.
Як лікувати та запобігти порфірії?
Лікування в основному залежить від типу порфірії та може бути досить успішним. Запобіжні заходи, які включають утримання від алкоголю та деяких ліків, є теж дуже важливими для тих, у кого діагностована спадкова порфірія, навіть тоді, якщо симптоми не проявляються. Пацієнти із порфірією, незалежно від вираженості проявів захворювання, повинні бути проінформовані про запобіжні заходи.
Чи є порфірія прогресуючим захворюванням? Чи може вона стати причиною смерті?
На прояви та перебіг захворювання суттєво впливає тип успадкованого аномального гена та фактори навколишнього середовища. Ці чинники в більшості випадків мають більший вплив, ніж успадкований ген сам по собі. Ризик важких ускладнень або навіть смерті при гострій порфірії часто зменшується, коли особи із цим захворюванням обізнані із діагнозом та застосовують відповідні профілактичні заходи. Навіть із сучасним лікуванням та запобіганням, деякі пацієнти страждають від повторних нападів хвороби. Однак, прогресивне погіршення та смерть від порфірії в наш час є досить рідкісним явищем.
Чи завжди шкідливе сонячне світло?
Підвищена чутливість до світла (фотосенсибілізація) властива практично усім формам порфірії, проте ступінь вираженості її коливається в широких межах. Пацієнти із чутливістю до дії сонячного світла мають високий рівень порфірину у плазмі крові, який, залежно від типу порфірину, виникає у печінці або кістковому мозку. Ультрафіолетові промені, вступаючи у взаємодію із порфірином, пошкоджують тканини шкіри. Деякі види лікування можуть допомогти пацієнтам переносити дію сонячних променів, навіть не впливаючи на рівень порфірину. У деякий випадках лікування спрямоване на зниження рівня порфірину, що дозволяє переносити дію сонячного світла.
Чи варто повідомляти лікаря, що пацієнт хворий на порфірію, навіть якщо вона латентна?
Так! Діагноз порфірії є досить важливою медичною інформацією, навіть тоді, коли порфірія латентна. Це може, наприклад, впливати на вибір ліків та інших лікувальних процедур, вибір анестезії при хірургічному втручанні або рекомендації щодо дієти.
Чи існує ризик при хірургічному втручанні та під час вагітності?
Знову ж таки, це залежить від типу порфірії. Хірургічне втручання може збільшити ризик нападів при гострих формах порфірії. Цей ризик можна значно зменшити, якщо вдатися до певних запобіжних заходів. Ваш хірург та/або анестезіолог повинен проконсультуватися із вашим лікуючим лікарем перед госпіталізацією для хірургічного втручання. Такі консультації є також бажаними під час вагітності. Хоча напади гострої порфірії можуть мати місце під час вагітності, вони є набагато рідшими, ніж сподівані. Лікування гострої форми порфірії під час вагітності є досить ефективним.
Короткий огляд усіх типів порфірії
ALAD Порфірія (ADP). Ця форма порфірії успадковується по аутосомно-рецесивному типу і є досить рідкісним захворюванням. Симптоми дуже схожі на ті, які є при гострій періодичній порфірії. Це захворювання характеризується дефіцитом в організмі ферменту дегідратази дельта-амінолевуліновоїї кислоти (ALAD) та підвищеним вмістом в сечі дельта-амніолевулінової кислоти (ALA).
Гостра інтермітуюча порфірія (ГІП). Це є одна із вроджених печінкових порфірій. Тип успадкування – аутосомно-домінантний. Захворювання характеризується дефіцитом ферменту порфобіліногендезамінази (ПБГД), також відомим як гідрометилбілансинтетаза. Дефіцит лише цього одного ферменту клінічно проявляється при дії провокуючих факторів, як от гормони, ліки, різкі зміни дієти. Іноді не вдається їх чітко ідентифікувати.
Симптоми. Часто перші прояви ГІП з’являються у періоді статевого дозрівання, особливо у жінок, на фоні гормональних порушень. Симптоми проявляються у вигляді повторних нападів гострого болю у животі, нудоти, блювоти, запорів, болю у спині, руках та ногах, слабкості м’язів, затримки сечовиділення, сильного серцебиття, збудження, галюцинацій, іноді судомних нападів. Часом рівень солі (натрій хлориду) в крові значно збільшується і впливає на деякі із перерахованих симптомів. Шкіра не вражається.
Діагностика складна, оскільки це захворювання є досить рідкісним і його симптоми дуже схожі на ознаки інших хвороб. З іншого боку, діагноз ГІП та інших типів порфірії ставиться помилково тим особам, які її не мають взагалі, особливо якщо тести проводяться або інтерпретуються невірно. Виявлення високого рівня дельтаамінолевулінової кислоти (ALA) та порфобіліногену в сечі підтверджує наявність однієї із гострих порфірій. Якщо рівень ПБГД є зниженим у еритроцитах, то встановлюють діагноз гострої періодичної порфірії. Однак нормальні значення ПБГД у еритроцитах не виключають діагнозу ГІП, оскільки не у всіх пацієнтів із ГІП спостерігається дефіцит ензиму.
Якщо відомо, що у когось із родини вже встановлювався діагноз гострої періодичної порфірії та рівень ферментів у крові знижений, то і в інших членів родини, які можливо успадкували дефіцит ПБГД, теж потрібно провести аналізи для визначення рівня ферментів у кров’яних клітинах на підтвердження чи спростування діагнозу гострої періодичної порфірії. Якщо порфірія латентна, то встановлення діагнозу дозволить запобігти нападам шляхом уникнення провокуючих факторів. Однак, у членів деяких родин, де траплялись випадки гострої періодичної порфірії, рівень ПБГД може бути нормальним у еритроцитах, але низьким лише в печінці та інших тканинах. Псевдопозитивні результати аналізів можливі при порушенні правил забору та транспортування.
ДНК – це структура в усіх живих клітинах, в якій закодована вся генетична інформація особи. Багато різноманітних мутацій було визначено на ділянці ДНК, яка включає ген ПБГД. Майже кожна сім’я із гострою періодичною порфірією має інший тип мутації цього гена. Однак, в межах однієї родини, мутація є однаковою для усіх її членів. Надзвичайно важливим є встановлення, яка саме мутація поширена в певній родині, оскільки це знання дає змогу визначити носіїв гену гострої періодичної порфірії за допомогою ДНК-діагностики. Цей підхід є набагато точнішим, ніж визначення активності ферменту ПБГД у червоних кров’яних клітинах. Однак, на сьогодні ДНК діагностика порфірії можлива лише в деяких спеціалізованих дослідних лабораторіях.
Лікування та прогноз. Госпіталізація необхідна у випадках гострих нападів хвороби. Зазвичай призначають ліки, щоб подолати біль, нудоту та блювоту. В таких випадках необхідний постійний нагляд лікаря.
Прийом великої кількості глюкози та інших вуглеводів може полегшити перебіг нападів. Їх слід приймати орально або внутрішньовенно при незначній гостроті процесу. Внутрішньовенна гемотерапія є більш дієвою в подоланні нападів хвороби. Її можна розпочинати після лікування глюкозою. Однак, кращі результати гемотерапії досягаються, якщо вона розпочинається на початкових стадіях нападів порфірії.
Під час лікування нападів порфірії варто контролювати баланс солі та води в організмі. Слід припинити вживання небезпечних ліків: барбітуратів, сульфонамідів та інших. Напади можуть провокуватись гіпокалорійною дієтою з обмеженою кількістю вуглеводів. Тому консультації щодо дієти є дуже важливими. Передменструальні напади найчастіше швидко послаблюються з початком місячних. Вживання гормонів може попередити ці напади.
Вчасно недіагностована ГІП є небезпечним захворюванням!
Прогноз при ГІП є зазвичай сприятливий, якщо лікування та запобіжні заходи розпочаті задовго до важких нервових пошкоджень. Не дивлячись на те, що симптоми зникають після нападів хвороби, у деяких хворих розвивається хронічний біль. Нервові ураження та м’язева слабкість можуть виникати знову і знову через деякий час після важких нападів хвороби. Ускладнення, пов’язані із розумовою діяльністю, можуть виникати під час нападів, але зазвичай не переходять у хронічні.
Дієта. Усі пацієнти із гострою інтермітуючою порфірією, які схильні до нападів цього захворювання, повинні вживати достатню, або дещо збільшену кількість вуглеводів та калорій. Обов’язкова консультація дієтолога із розрахунком індивідуальної дієти у випадку схуднення.
Діти. Вагітність переноситься значно краще, ніж раніше вважалося. Нащадки мають 50% ризик успадкування гена ГІП, але у більшості випадків порфірія залишається латентною протягом усього життя. Розроблені ефективні лікувальні та профілактичні заходи. З огляду на це, пацієнти із порфірією можуть мати дітей так само, як і усі інші.
Вроджена еритропоетична порфірія (ВЕП) – рідкісний аутосомно-рецесивно успадковуваний варіант порфірії, відомий також, як хвороба Хантера (Gunther’s disease). У різних родинах було встановлено різноманітні мутації гена цього фермента. Характерною особливістю ВЕП є те, що симптоми проявляються ще в ранньому віці. Іноді ознакою цього типу порфірії є анемія плоду ще до народження. У менш складних випадках симптоми можуть виникнути у зрілому віці. Рівень порфірину значно збільшений у кістковому мозку, червоних кров’яних клітинах, плазмі, сечі та калі. Порфірин також відкладається в зубах та кістках.
Симптоми. Підвищена чутливість до сонячного світла призводить до опіків, рубців та збільшення оволосіння, особливо відкритих ділянок шкіри (обличчя, долоні). Часто нашаровується бактеріальна інфекція. В результаті скороченого терміну функціонування червоних кров’яних клітин розвивається анемія, а також компенсаторно пришвидшується синтез гему та гемоглобіну.
Лікування та прогноз. Переливання крові та, можливо, видалення селезінки можуть зменшити продукування порфірину у кістковому мозку. Іноді досить дієвим є вживання активованого вугілля. Деяким пацієнтам значно допомогла трансплантація кісткового мозку. Трансплантація стовбурових клітин та генна терапія, можливо, використовуватимуться в майбутньому.
Пізня шкірна порфірія (ПШП). Цей тип порфірії, що є результатом дефіциту ферменту уропорфіриногендекарбоксилази зустрічається найчастіше. Взагалі-то це набута хвороба, але деякі особи мають її генетичну (аутосомно-домінантну) або сімейну форму. У більшості випадків спадкова порфірія залишається латентною до кінця життя і не виявляє жодних симптомів.
ПШП є печінковою порфірією. При загостренні у клітинах печінки накопичується велика кількість порфірину. Провокуючими факторами розвитку ПШП є: залізо, алкоголь, вірусний гепатит С, ВІЛ інфекція, естрогени (які вживаються орально з метою контрацепції та при лікуванні раку простати), куріння, що призводить до нестачі ферменту уропорфіриногендекарбоксилази в печінці, гемохроматоз.
Симптоми ПШП зазвичай пов’язані із шкірою. На ділянках, які найбільше зазнають дії сонячного світла – руках та обличчі – з’являються опіки. Шкіра тут легко пошкоджується: іноді темніє та потовщується, значно збільшується оволосіння. Порушення роботи внутрішніх органів та нервової системи не є характерними для РСТ. Характерними є незначні порушення функції печінки. В деяких випадках розвивається цироз печінки і навіть рак.
Діагностика. Найкращим скринінговим тестом для ПШП є вимірювання рівня порфірину в плазмі. Це може відрізнити ПШП від пістрявої порфірії. Похідні порфірину в сечі (переважно уропорфірин та 7- карбоксилат порфірину) та калі (переважно ізокопропорфірин) допомагають підтвердити діагноз. Наявність спадкового дефіциту уропорфіриногендекарбоксилази може бути встановлена визначенням кількості ферментів в еритроцитах. Така особливість є характерною для 20% пацієнтів із РСТ.
Лікування та прогноз. Лікування ПШП є досить дієвим при сімейній та несімейній порфірії. Слід уникати провокуючих факторів. Найбільш дієвим способом лікування є повторні кровопускання крові з метою зменшення рівня заліза в крові. Така процедура фактично зменшує рівень заліза в усьому організмі. Зазвичай видалення 300 мл крові щотижня протягом 1 місяця є достатнім, що підтверджує невисокий рівень заліза в організмі більшості пацієнтів із ПШП. Контрольним тестом є рівень феритину та порфірину в плазмі. Кровопускання припиняють, коли рівень феритину зменшується до 20 нг/мл. Інший підхід до лікування полягає у дозованому вживанні або хлорохіну (125 мг двічі на тиждень), або гідроксохлорохіну (100 мг двічі на тиждень). Звичайна доза цих препаратів не застосовується, оскільки в цьому випадку вона може стати причиною ушкодження печінки та посилення фотосенсибілізації пацієнтів із ПШП. Рецидиви трапляються відносно рідко, у кропусканнях при тривалих ремісіях немає потреби.
Гепатоеритропоетична порфірія (ГЕП). Цей досить рідкісний тип порфірії теж характеризується дефіцитом уропорфіріногендекарбоксилази. Нестача ферменту успадковується аутосомно-рецесивно. Ознаки, що вказують на ГЕП дуже схожі на ті, що і при ВЕП (вроджена еритропоетична порфірія), однак опіки шкіри проявляються вже в ранньому віці. Рівень порфірину значно збільшений в кістковому мозку, червоних кров’яних клітинах, печінці, сечі та калі.
Спадкова копропорфірія (СКП). Це аутосомно-домінантна форма печінкової порфірії, що має подібні ознаки до ГІП. Єдиною відмінністю є те, що у деяких пацієнтів розвивається надчутливість до сонячного світла. “Дефіцитний” фермент – копропорфіріногеноксидаза. Діагноз підтверджується виявленням копропорфірину (особливо III типу) в сечі та калі. Рівень інших типів порфірину значно не збільшується. Рівень сечових ALA та PBG підвищується під час нападів хвороби, але після відповідного лікування нормалізується.
Для визначення кількісті ферменту червоні кровяні тільця не використовуються, оскільки фермент знаходиться у мітохондріях, яких немає у червоних кровяних тільцях. Профілактика та лікування нападів хвороби такі самі як і при ГІП.
“Пістрява” порфірія. Ця форма печінкової порфірії є найбільш поширеною серед населення Південної Африки, рідко трапляючись в інших місцевостях. Успадковується аутосомно-домінантно. Характерними є напади та надчутливість до сонячного світла. “Дефіцитний” фермент – копропорфіріногеноксидаза. Діагноз підтверджується виявленням копропорфірину та протопорфірину у фекаліях. Найчутливішим методом скринінгу “пістрявої” порфірії є визначення ферменту в плазмі. У пацієнтів, які мають проблеми із шкірою, потрібно точно відрізнити “пістряву” порфірію чи СКП від ПШП, оскільки флеботомія чи малі дози хлорохіну не дають бажаного результату при “пістрявій” порфірії та СКП. Важкі напади хвороби лікуються так само, які і при ГІП.
Еритропоетична протопорфірія або протопорфірія. Цей вид порфірії розвивається внаслідок дефіциту ферохелатази. Хвороба успадковується за аутосомно-домінантним типом. У різних сім’ях із протопорфірією були визначені різноманітні мутації гену ферохелатази. Протопорфірин акумулюється у кістковому мозку, червоних кров’яних тільцях та іноді у печінці. Надлишок протопорфірину виділяється печінкою у жовч, після чого він потрапляє у кишківник. Таким чином протопорфірин виявляється у фекаліях. Рівень порфірину в сечі є нормальним. Діагноз підтверджується наявністю надлишку протопорфірину у плазмі, фекаліях та червоних кров’яних тільцях.
Симптоми. Набряки, опіки, свербіння та почервоніння шкіри після нетривалого перебування під дією сонячного світла (навіть через віконне скло) – характерні ознаки протопорфірії. Як правило, ці ознаки зникають через 12-24 години без особливого лікування. Шкіра не змінює кольору, немає рубців. Часом проблеми із шкірою виникають лише після тривалого перебування на сонці. Ураження шкіри можуть перейти у хронічні, що потребує спеціального лікування. Іноді на тілі залишаються рубці. Ураження шкіри менш виражені, ніж при ПШК. Проблеми із шкірою, як правило, з’являються у дитячому віці. Вони загострюються влітку, протягом життя можуть повторюватись неодноразово. Іншими ознаками протопорфірії є наявніть жовчного каміння, часом важкі порушення функції печінки. У деяких носіїв гену еритропоетичної протопорфірії симптоми хвороби можуть і не проявлятися, рівень порфірину залишається нормальним.
Лікування та прогноз. Лікування бета-каротином зменшує чутливість до сонячного світла, але не впливає на загальний рівень порфірину. Люмітен є єдиним фармацевтичним препаратом бета-каротину, який рекомендують при еритропоетичній протопорфірії. У деяких випадках ін’єкції холестираміну можуть знизити рівень порфірину.
У пацієнтів із еритропоетичною протопорфірією протопорфірин може потрапляти в печінку, призводячи до розвитку печінкової недостатності. На щастя, таке трапляється дуже рідко.
Неврологічні проблеми не характерні для хворих на еритропоетичну протопорфірію. Хоча, деякі пацієнти все ж мали неврологічні симптоми схожі на ті, що і при гострих порфіріях. Для запобігання ускладнень варто уникати препаратів, що призводять до холестазу, включаючи естрогени та алкоголь. Деякі пацієнти із еритропоетичною протопорфірією повідомляли, що вживання алкогольних напоїв значно підвищує світлочутливість.
Безпечні та небезпечні ліки
Нижче поданий список лише деяких лікарських препаратів, які можна вживати без застережень при гострій інтермітуючій порфірії, “пістрявій” порфірії, спадковій копропорфірії та ALAD порфірії, та тих, що є небезпечними для пацієнтів із порфірією усіх типів.
Ліки, застосування яких є шкідливим для пацієнтів із порфірією:
- барбітурати;
- сульфонамідні антибіотики;
- багато інших транквілізаторів та седативних препаратів (мекробамат, глютамідин);
- грізеофулін;
- протиепілептичні засоби;
- контрацептиви;
- алкоголь;
- ерготаміни;
- метоклопрамід;
- ріфампіцин;
- диклофенак;
- даназол.
Перелік препаратів, вживання яких є безпечним для пацієнтів із порфірією:
- наркотичні анальгетики (морфій, меперідин, кодеїн, та ін.);
- аспірин та ацетамінофени;
- фенотіазіни;
- пеніцилін та його похідні;
- хлоргідрати;
- стрептоміцин;
- глюкокортикоїди;
- броміди;
- інсулін;
- атропін;
- циметидин;
- антидепресанти.
Рекомендації щодо лікарських препаратів базуються на досвіді пацієнтів із порфірією, які мали ускладнення внаслідок вживання тих чи інших ліків, та на основі тестувань, які проводились на дослідних тваринах з порфірією. Якщо ж виникає питання необхідності вживання того чи іншого препарату, обов’язкова консультація лікуючого лікаря.
Діагностичне тестування порфірії
Існує багато доступних тестів на наявність порфірії, однак, часто досить важко визначити, який із них варто спробувати. Більшість із цих тестів є досить дорогими, складними у трактуванні. Тести мають різну чутливість та специфічність. Якщо тест достатньо чутливий, малоймовірно, що результат буде псевдонегативним (тобто не діагностує порфірію у пацієнта, який дійсно її має). Якщо ж тест є специфічним, то малоймовірно, що результат буде псевдопозитивним (тобто вкаже про наявність порфірії у здорової людини). Деякі тести є і чутливими і специфічними для пацієнтів із симптомами, схожими на симптоми порфірії. При підозрі на гостру порфірію слід провести аналіз сечі на дельта-амніолевулінову кислоту та порфобіліноген. Якщо ж з’являються ознаки хвороби, що пов’язані із шкірою, найефективнішим методом встановлення чи підтвердження діагнозу є визначення порфірину в плазмі. Якщо хоча б один із цих тестів є позитивним, бажаним є проведення усіх інших. Аналіз сечі, калу або червоних кров’яних тілець на наявність порфірину в них не є досить інформативними для початкового скринінгу через низьку точність. Визначення ферментів синтезу гему застосовується лише в тих випадках, коли порфірія вже зустрічалась у членів певної родини. Усі тестування та аналізи слід проводити в спеціальних лабораторіях, де цим займаються спеціалісти, які можуть правильно пояснити результати тих чи інших досліджень.
Якщо ж тестування проводилось не в спеціальній лабораторії, то перед остаточною постановкою діагнозу слід обов’язково проконсультуватися із спеціалістом.
Переглянуто редакційною колегією I.B.I.S.: 29/11/2002
Поренцефалія
(Porencephaly)
Світлана Зраєвська
Лікар акушер-гінеколог
Костопільської центральної районної лікарні Рівненської області
Визначення:
Терміном „поренцефалія” (синоніми: поренцефалічна киста, шизенцефалія, вроджене розщеплення мозку) визначають внутрішньочерепні кистозні порожнини, в яких міститься спинномозкова рідина. Вони можуть поєднуватись або не поєднуватись з шлуночковою системою мозку і субарахноїдальним простором.
Частота:
Поренцефалія відноситься до захворювань, що рідко зустрічаються. Приблизно в 25 із 1000 дітей з ураженням головного мозку.
Етіологія і патогенез:
Поренцефалію поділяють на дві групи: справжню і псевдопоренцефалію.
Справжня поренцефалія являє собою аномалію розвитку, в основі якої лежить порушення міграції клітин, що беруть участь у формуванні кори півкуль головного мозку, внаслідок чого утворюються вогнищеві дефекти як сірої, так і білої речовини головного мозку. В зв’язку з відсутністю нервової тканини субарахноїдальний простір збільшується, заповнюючи пустоти, що супроводжується утворенням порозних кист. Термін „порозна” означає часто зустрічну комунікацію мозку з субарахноїдальним простором.
Псевдопоренцефалія є результатом вогнищевої деструкції паренхіми мозку через дію судинних, інфекційних і травматичних факторів, що впливають на плід чи на новонародженого. Прикладом набутої поренцефалії можуть бути кисти, що утворились у новонароджених дітей з гідроцефалією після повторних пункцій шлуночкової системи мозку до проведення шунтування в ранньому неонатальному періоді.
Порушення цитоархітектоніки, пов’язані зі змінами міграції клітин, включають в себе широкий спектр патології до поренцефалії. У першому випадку порушення міграції виражені в меншій мірі, звивини мозку зменшені. Патологія прогресує через великі небагаточисленні звивини мозку, ліссенцефалію чи агірію (відсутність звивин мозку) до поренцефалії, при якій зміни настільки виражені, що в процес втягуються ділянки і кори, і білої речовини.
Патологічна анатомія:
Для поренцефалії типові кистозні порожнини, різні за розміром, локалізовані біля сільвієвої борозни, частіше симетричної форми. Нерідко вони поєднуються з іншими порушеннями цитоархітектоніки, такими, як мікрогірія чи гетеротопія сірої речовини. Мозолясте тіло може бути гіпоплазованим або відсутнім.
Псевдопоренцефалія від справжньої відрізняється тим, що процес майже завжди однобічний і супроводжується ознаками загальних та ішемічних порушень. Справжня поренцефалія часто поєднується з мікроцефалією. Вентрикуломегалія супроводжує як справжню, так і псевдопоренцефалію, і часто буває симетричною. Незважаючи на те, що поренцефалічні кисти часто локалізуються у великих півкулях, вони можуть бути у мозочку та спинному мозку.
Діагностика:
Діагностика ґрунтується на виявленні внутрішньочерепних кист, які при справжній поренцефалії можуть бути двобічними і рідше однобічними. Звертає на себе увагу розширення шлуночків і зміщення серединної лінії. Цінну інформацію можна отримати при дослідженні вінцевої площини, коли чітко визначаються дефекти мозкової тканини.
Диференційна діагностика:
Диференційна діагностика проводиться з вродженим кистозом мозку, наприклад, арахноїдальними кистами і кистозними пухлинами. Локалізація кистозних утворів в основі черепа нетипова для поренцефалії. Відповідно, в таких випадках в першу чергу слід передбачити ймовірність арахноїдальних кист та інших пухлин. Позитивний діагноз може бути поставлений в більшості випадків при виявленні обширного пошкодження півкуль мозку.
Прогноз:
Велике значення для прогнозу має розмір ураження. Наслідки для дітей із справжньою поренцефалією вкрай несприятливі, у всіх випадках наступають виражені інтелектуальні та неврологічні порушення. Виникає спастична тетраплегія та сліпота.
Акушерська тактика:
До періоду життєздатності плоду рекомендується переривання вагітності. Після досягнення життєздатності – консервативне родорозрішення.
Номер з каталогу МІМ:
175780 Brain small vessel disease 1 with or without ocular anomalies; BSVD1.
Література:
- Gorlin RJ, Cohen MM, Hennekam RCM. Syndromes of the Head and Neck. Oxford University Press. 2001:17,176,832,1020,1079.
- Warkany J. Congenital Malformations. Notes and Comments. Year Book Publisher. Chicago. 1981:247-251.
- Пренатальная диагностика врожденных пороков развития плода /Р. Ромеро, Дж. Пилу, Ф. Дженти, А. Гидини, Дж. С. Хоббинс.- М.: Медицина,1994.- C. 58-61.
- Тератология человека /Под ред. Г.И. Лазюка.- М.: Медицина,1979.- C. 108.
Переглянуто редакційною колегією I.B.I.S.: 11/09/2003
Довжина тіла близнюків при народженні
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”
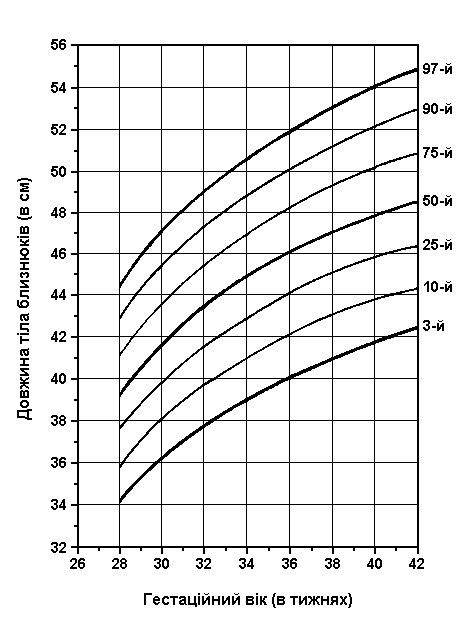
Довжина тіла близнюків при народжені (3-й, 10-й, 25-й, 50-й, 75-й, 90-й та 97-й персентилі) була визначена Leroy et al (Acta Genet Med Gemellol 31:199, 1982). Це французське дослідження 1049 близнюків, включало дані про тих близнюків, що народилися з гестаційним віком від 28 до 42 тижнів, та поєднувало дані без зазначення зиготності, статі та даних про народження. Довжина тіла близнюків починає відставати від довжини тіла плодів в одноплідній вагітності приблизно з 30 тижня, це відставання зберігається до самого моменту народження.
Синдром полікістозних яєчників
(Polycystic Ovary Syndrome)
Л.В. Дудра
Акушер-гінеколог
Волинського обласного центру планування сім’ї та репродукції людини
Основні діагностичні критерії:
Під синдромом полікістозних яєчників (СПКЯ) розуміють генетично зумовлений клінічний симптомокомплекс, що характеризується оліго- або аменореєю в поєднанні з ендокринними порушеннями. Менш постійними ознаками є гірсутизм, ожиріння, збільшення яєчників. Замість терміну “синдром полікістозних яєчників” часто використовується первинний термін “синдром склерокістозних яєчників”. Дане захворювання отримало назву згідно публікацій Stein і Leventhal – синдром Штейна-Левенталя, які описали 7 гірсутних жінок, що страждали непліддям, аменореєю і ановуляторною олігоменореєю. По даним гістологічного дослідження були виявлені множинні кісти субкортикального шару яєчників, потовщення білкової оболонки з фіброзом підлеглих тканин і велика кількість атретичних фолікулів з потовщенням theca interna.
Частота:
Згідно даних літератури, частота синдрому полікістозних яєчників складає від 1,8 до 11% серед гінекологічних хворих, серед неплідних жінок – від 0,6% до 4,3% жінок.
Етіологія:
Полікістоз яєчників розглядається як фенотипічне вираження різноманітних хромосомних конституцій, що в свою чергу визначає гетерогенність клінічних проявів синдрома. Дерматогліфічні обстеження підтвердили дані літератури про певну роль спадкового фактора у виникненні даної патології. Передача здійснюється через Х-зчеплену домінантну трансмісію. Існує думка, що захворювання може успадковуватись не лише по жіночій, а й по чоловічій лінії.
Патогенез:
Синдром полікістозних яєчників – це генетично обумовлене захворювання, в основі якого лежить порушення синтезу статевих гормонів внаслідок неповноцінності ензимних систем.
Згідно однієї з поширених гіпотез в основі захворювання лежить первинний, генетично зумовлений дефіцит ферментів яєчника. Найчастіше зустрічається порушення одного із заключних етапів стероїдогенезу – на рівні 19-гідроксилази: недостатність цього ферменту блокує перетворення андрогенних попередників в естрогени. Інший ферментний дефект – недостатність 3-бета-дегідрогенази; в даному випадку біосинтез стероїдів приводить до збільшення рівня дегідроепіандростерона і андростендіона і значного підвищення тестостерону. Порушення біосинтезу стероїдів в яєчниках обумовлює (по механізму зворотнього зв’язку) дизфункцію гіпоталамо-гіпофізарної системи, що виявляється в порушенні циклічної секреції гонадотропінів і в свою чергу приводить до гіперплазії строми, тека-тканини і клітин воріт яєчників на фоні ферментного дефекту стероїдогенезу. В сучасній гінекологічній ендокринології існує чітке уявлення про найбільш поширені форми даного захворювання. До них слід віднести типову форму синдрома полікістозних яєчників з оваріальною гіперандрогенією, в певній мірі відповідаючій власне синдрому Штейна-Левенталя; поєднану форму захворювання, яка розвивається на фоні яєчникової і наднирникової гіперандрогенії, а також центральну форму з вираженими порушеннями зі сторони гіпоталамо-гіпофізарної системи.
Аналіз співвідношень гонадотропних та статевих гормонів і моноамінів в біологічних рідинах у хворих підтверджує гетерогенність даної патології. При аналізі виявляються 4 варіанта співвідношень між ними. Перший – висока продукція лютеінізуючого гормону (ЛГ) при нормальному рівні фолікулостимулюючого гормону (ФСГ) і пролактину; співвідношення ЛГ/ФСГ значно збільшено (до 5), концентрація дофаміну і серотоніну в межах норми; рівень естрогенів достатній. Подібні явища можливі при первинному пораженні яєчників і збереженні гіпоталамо-гіпофізарних взаємовідносин. Другий варіант – рівень ЛГ в плазмі не такий високий, рівень ФСГ знижений, їх співвідношення 3,2. Екскреція дофаміну і серотоніну підвищена, 17-КС і ДЕА також підвищена, проба з дексаметазоном позитивна. Розвиток захворювання пов’язують з одночасним пораженням кори наднирників та яєчників. Для третього варіанту патогномонічною ознакою є незначне збільшення ЛГ і зниження ФСГ, співвідношення ЛГ/ФСГ в 4-5 раз вище норми. Вміст дофаміну знижений, серотоніну підвищений, 17-КС і ДЕА в межах норми. Дана картина характерна для первинного порущення центральних моноамінергічних механізмів. Для четвертого варіанту характерне зниження дофаміну і підвищення серотоніну, збільшення секреції ЛГ, ФСГ, пролактину і андрогенів, відмічається порушення механізмів оберненого зв’язку між андрогенами і пролактином. Високий рівень пролактину підтримує гіперандрогенію.
Клініка:
Синдром склерополікістозних яєчників частіше виникає в періоді статевого дозрівання, іноді у віці 20-30 років.
Синдром полікістозних яєчників супроводжується порушеннями менструального циклу, ступінь важкості яких обумовлений характером дефекту ферментних систем яєчників. Важкість цих порушень різноманітна: олігоменорея або вторинна аменорея. Олігоменорея трансформується послідовно в ациклічні маточні кровотечі. Порушення менструального циклу супроводжується хронічною ановуляцією, яка приводить до функціонального непліддя, частіше первинного.
Гірсутизм проявляється в рості волосся на обличчі, біля навколососкових кружків, на передній черевній стінці, підсилений ріст волосся на кінцівках. З інших клінічних проявів зустрічається ожиріння в поєднанні з симптомами гіпоталамо-гіпофізарних порушень: розтягнення на животі та бедрах, підвищення внутрішньочерепного тиску.
Деякі автори відмічають при синдромі полікістозних яєчників у підлітків наявність гіперінсулінізму, ліподистрофії, папілярно-пігментної дистрофії шкіри.
При ультразвуковому скануванні яєчників пацієнтів зі СПКЯ виявляються, згідно критеріїв Адамса (1996):
- Збільшені розміри (об’єм більше 10 см3 або мінімальний розмір більше 4 см);
- Більше 15 округлих анехогенних утворів діаметром від 3-8 мм, розташованих під капсулою яєчника;
- Гіперехогенність строми яєчника.
Поєднана форма оваріальної і наднирникової гіперандрогенії характеризується більш пізнішим менархе. Порушення менструального циклу протікають по типу вторинної аменореї, рідше олігоменореї. У хворих з’являється надлишковий ріст волосся на обличчі, кінцівках, тулубі. Тип будови тіла подібний до інтерсексуального. По тестам функціональної діагностики виявляються ановуляція і низька естрогенна насиченість. При ультразвуковому обстеженні органів малого таза визначається матка розмірами менше норми, яєчники збільшені.
У більшості хворих з центральною формою синдрому батьки чи ближні родичі страждали функціональними або органічними захворюваннями нервової системи. В анамнезі часто вказується на високий інфекційний індекс, в тому числі перенесені нейроінфекції, психічні травми, травми черепа. Крім порушень менструального циклу та непліддя, хворих турбують головні болі, погана пам’ять, швидка втомлюваність, слабкість, патологічна добавка у вазі.
Діагноз:
Діагноз синдрому полікістозних яєчників ставиться на основі клінічної картини, для якої характерні: ановуляторні порушення менструального циклу, відсутність вагітності у молодих жінок, ожиріння і помірно виражений гірсутизм при збереженні жіночого фенотипу.
Диференціальний діагноз:
Характерними для цього синдрому зміни в яєчниках можна знайти також при різних патологічних станах: адреногенітальному синдромі, синдромі Іценко-Кушинга, гіперпролактинемії, генітальному туберкульозі, на фоні постійних рецидивуючих запальних захворювань, перенесених в пубертатному віці або в ранній репродуктивній фазі. Провокуючими моментами можуть бути стресові ситуації.
Лікування:
Підходи до лікування СПКЯ залежать від репродуктивної мети пацієнтки. Ведучою лінією лікування повинно стати прагнення до досягнення овуляції. Лікування кломіфеном у відповідності зі стандартним протоколом індукції овуляції є розумним початковим підходом і дозволяє на протязі 6 циклів викликати овуляцію у 80% і добитись вагітності у 40% пацієнтів. Препарат назначають пацієнткам з базовим рівнем Е<50 пг/мл з 3-5 дня менструального цикла в дозі 50-100 мл/д на протязі 5 днів. Овуляторна доза хоріонічного гонадотропіна складає 5000-10000 МЕ на 13 день при наявності по даним УЗД фолікула >8 см. У кломіфенрезистентних пацієнток альтернативним методом є застосування гонадотропінів:
- Людський менопаузальний гонадотропін (пергонал, хумегон, меногон);
- Людський ФСГ;
- Високочистий метродін (метродін ВЧ);
- Рекомбінантний ФСГ (пурегон, гонал-Ф);
Призначення їх здійснюється за індивідуальною схемою, в якій доза гонадотропіна залежить від відповіді яєчника в цьому ж циклі.
Альтернативним підходом при кломіфенрезистентності є хірургічне лікування з використанням методу лапароскопії. Ціль операції – руйнування тканини строми яєчника, розташованої центрально, оскільки саме в стромі знаходяться секретуючі андрогени клітини. При зменшенні об’єму цієї тканини рівень андрогенів в крові знижується, що веде до розриву замкненого кола патогенезу і часто до відновлення овуляторних циклів. В процесі удосконалення хірургічного лікування зупинились на необхідності розширеної клиновидної резекції яєчників з частковою демедуляцією. Способи хірургічного лікування СПКЯ із застосуванням лапароскопічного доступу:
- Множинна пункція дрібних кіст яєчників;
- Множинна перфорація яєчників дерматомом;
- Множинна електропунктура яєчників;
- Множинні діатермокоагуляційні насічки;
- Двостороння клиновидна резекція яєчників та інші.
Профілактика:
Дівчатка, що народились від матерів, які хворіли на синдром полікістозних яєчників, успадковують схильність до порушень репродуктивної системи. Вони потребують нагляду дитячого гінеколога з метою раннього виявлення і своєчасної корекції порушень репродуктивної системи в періоді статевого дозрівання. Жінки, які страждають на синдром полікістозних яєчників, складають групу високого ризику по формуванню гіперпластичних процесів в репродуктивній системі і потребують систематичного лікарського контролю.
Номер з каталогу МІМ:
184700 Polycystic ovary syndrome 1; PCOS1.
Література:
- Гаспаров А.С., Кулаков В.Н. Роль лапароскопии в диагностике болезни поликистозных яичников (БПКЯ) и сопутствующей патологии органов малого таза //Проблемы репродукции.- 1995.- №2.- С.34-35.
- Грищенко В.І., Щербина М.О., Бережна Т.А. і співавт. До питання патогенезу та лікування синдрому полікістозних яєчників //ІПАГ.- 1998.- №5.- С.95-99.
- Дзенис Н.Г., Бухаров В.А., Фанченко Н.Г., Бухаров В.А., Фанченко Н.Д., Дуринян Э.Р., Назаренко Т.А., Сотникова Е.И. Стероидогенез в надпочечниках при синдроме поликистозных яичников //Проблемы репродукции.- 1997.- №3.- С.18-21.
- Іванюта Л.І., Акопян А.З. Хірургічне лікування синдрому полікістозних яєчників //Педіатрія, акушерство та гінекологія.-1998.- №4.- С.97-99.
- Іванюта Л.І., Беліс Н.І., Іванюта С.О. Доброякісні пухлини яєчників (огляд літератури) //Перинатологія та педіатрія.- 1999.- №1.- С.35-40.
- Іванюта Л.І., Іванюта С.О.Синдром передчасного виснаження яєчників та неплідність.//Педіатрія, акушерство та гінекологія .- 1998.- №1.- С.12-13.
- Кирющенков А.П., Совги М.Г. Поликистозные яичники //Акушерство и гинекология.- 1994.- №1.- С.11-14.
- Кузнецова И.В., Стрижаков А.Н. Роль гипоталамического синдрома периода полового созревания в патогенезе поликистозных яичников //Акушерство и гинекология.- 1996.- №2.- С.7-9.
- Мухина Н.Б., Геворкян М.А. Синдром поликистозных яичников //Проблемы репродукции.- 1999.- №6.- Т.5.- С.18-23.
- Paul D. Silva, Гладкова О.Н. Возможности стимуляции овуляции у больных с синдромом поликистозных яичников //Акушерство и гинекология.- 1997.- №3.- С.9-11.
- Adams J., Polson D.W., Franks S. Prevalence of polycystic ovaries in woman with anovulation and idiopathic hirsutism. British Medical Journal 1986;292(6543):355-359.
- Donesky D.W., Adashi E.Y. Surgically induced ovulation in the polycystiс ovary syndrome: wedge resection revisited in the age of laparoscopy. The Ares Serono Group.- Geneva: Switzerland. 1992:21-33.
- Franks S. Polycystic ovary syndrome. The New England Journal of Medicine 1995;333(13):853-861.
- Homburg R. Polycystic ovary syndrome – from gynaecological curiosity to multisystem endocrinopathy. Human Reproduction 1996;11(15):29-39.
- Hsuska J.W. A prospective study of polycystic ovarium. Fertility and Sterility 1984;52:891-893.
Переглянуто редакційною колегією I.B.I.S.: 15/11/2002
Вага близнюків при народженні (продовження)
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”
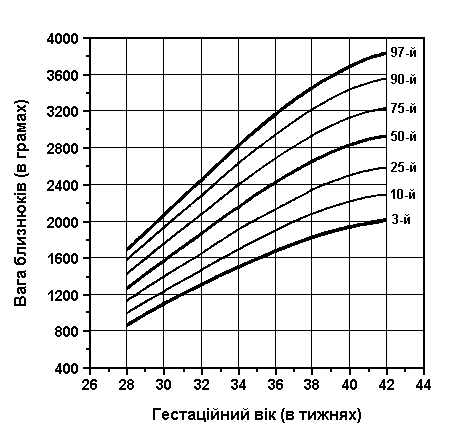
Вага близнюків при народжені (3-й, 10-й, 25-й, 50-й, 75-й, 90-й та 97-й персентилі) була визначена Leroy et al (Acta Genet Med Gemellol 31:199, 1982). Це французьке дослідження 1049 близнюків включало дані про тих близнюків, що народилися з гестаційним віком від 28 до 42 тижнів, та поєднані дані без зазначення зиготності, статі та даних про народження. Проте, ці дані свідчать, що діхоріонічні близнюки та новонароджені чоловічої статі є важчими за монохоріонічних близнюків та новонароджених жіночої статі. Вага близнюків починає відставати від ваги плодів в одноплідній вагітності приблизно з 30 тижня, це відставання зберігається до самого моменту народження.
Синдром Поланда
(Poland Syndrome)
О.М. Мислінчук
Лікар-генетик
Рівненський клінічний лікувально-діагностичний центр
Синдром описаний в 1841 році англійським патологом A. Рoland.
Виключення:
Однобічний дефект грудного м’язу, синдактилія кисті.
Основні діагностичні критерії:
Аплазія (чи часткова відсутність) великого грудного м’язу та синдактилія (чи олігосиндактилія) кисті на тому ж боці.
Клінічні ознаки:
Розрізняють 3 форми ураження кисті:
- Найчастіше синдактилія поєднується з брахімезофалангією. Олігодактилії немає, хоча фаланги II – IV пальців вкорочені та потовщені.
- Зустрічається також друга форма, коли дистальні фаланги ІІ-IV пальців відсутні.
- У 15% спостережень відмічається ектродактилія з дефектами п’ясткових кісток.
У більшості випадків рука вкорочена та рівномірно потоншена на всьому протязі.
Друга основна вада – аплазія грудино-реберної частини великого грудного м’язу, внаслідок чого грудна клітка на стороні ураження западає, чітко визначаються ребра та верхній зубчатий м’яз, який зазвичай прикривається m. pectoralis major. Молочна залоза відсутня чи гіпоплазована, у частині випадків виявляються додаткові соски або відсутність соска молочної залози. Вважається, що мікропроявами синдрому можуть бути стигми: аномалії дерматогліфіки, невеликі вкорочення кистей.
Асоційовані вади:
Вади внутрішніх органів не характерні, за винятком кількох описаних вад розвитку магістральних судин (коарктація аорти) і декстракардії. В окремих випадках виявляються килові випинання легеневої тканини в міжреберні проміжки, пахові та пупкові кили, гіпоплазія нирок, мікроцефалія і навіть потиличне енцефалоцеле. Але всі ці вади зустрічаються дуже рідко і їх не можна вважати характерними проявами синдрому.
Диференційний діагноз:
Схожі ураження кінцівок відмічаються при синдромі Мебіуса, для якого, крім того, характерні вроджені парези черепних нервів. На думку І.Herrmann і співавтора (1976) існує єдиний синдром Мебіуса-Поланда, а “аномалія Поланда” – лише один з компонентів цього стану. Скоріше за все, це два різних синдроми зі схожим морфогенезом.
Етіологія та патогенез:
До кінця невідомі. Аналізуючи виникнення синдрому, деякі автори мають думку, що різні неспецифічні за етіологією локальні порушення морфогенезу в ділянці великого грудного м’язу та прилягаючого до нього в цей період (44-48 доби ембріонального розвитку) зачатку кисті можуть порушити нормальний розвиток і кисті, і грудино-реберної частини великого грудного м’язу.
Інші дослідники пояснюють виникнення синдрому недостатнім кровопостачанням в басейні підключичної артерії.
Є вказівки на те, що значна частина вагітностей, від яких народились діти з синдромом Поланда, були небажаними та жінки приймали різні препарати для переривання вагітності.
Поширеність синдрому Поланда:
В популяції становить 1 випадок на 32000 народжень.
Співвідношення між статями: 3:2.
Вік прояву: пренатальний період.
Успадкування:
Переважаюча більшість спостережень синдрому Поланда спорадичні. Хоча відомо декілька описів синдрому у сибсів, нащадків хворих чи більш віддалених родичів. Скоріше за все, синдром Поланда гетерогенний. У деякій (невеликій) частині випадків він може бути звўязаний з генетично (мультифакторіально) детермінованим стенозом підключичної артерії. У більшості ж спостережень він виникає внаслідок неспецифічних за природою і негенетичних за етіологією локальних порушень морфогенезу.
Медико-генетичне консультування:
Якщо даних на користь генетичної детермінації немає, то прогноз вважається сприятливим.
Номер з каталогу МІМ:
173800 Poland Syndrome.
Літературні джерела:
- Jones KL. Smith’s Recognizable Patterns of Human Malformation. 5-th ed. 1981.
- Warkany J. Congenital malformation (notes and comments).- P. 962-963, 1025-1026.
- Наследственные синдромы и медико – генетическое консультирование: Справочник /Козлова С.И., Семанова Е.М., и др. – Л.: Медицина, 1987, C. 172-173.
- Тератология человека /Под. ред. Г.И. Лазюка. – М.: Медицина, 1979, C. 378 – 380.
Переглянуто редакційною колегією I.B.I.S.: 05/02/2002
Вага близнюків при народженні
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”
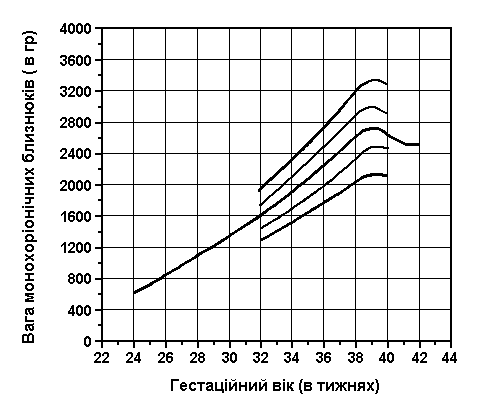
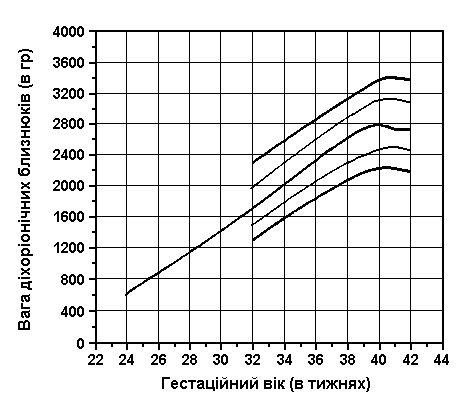
Дані про 10-й, 25-й, 50-й, 75-й та 90-й персентилі показників ваги базуються на обстеженні 701 монохоріонічного та 1748 діхоріонічних близнюків. Використовувались лише дані про живонароджених дітей без вроджених вад розвитку чи гемолітичної хвороби, які народились на східному узбережжі США. 10% з цих дітей не відносились до європеоїдної раси. Взято з Naeye et al (Pediatrics 37:409, 1966).
Планування сім’ї
(Family Planning)
Н.І. Юськів
Лікар-генетик
Волинського обласного дитячого територіального медичного об’єднання
Всі жінки дітородного віку в період планування вагітності повинні виконати декілька порад, які підвищать їхні шанси народити здорову дитину. Ці поради включають: щоденний прийом фолієвої кислоти (вітаміну групи “В”), ведення здорового способу життя, консультація лікаря та лабораторні обстеження.
Яким чином допомагає щоденний прийом фолієвої кислоти?
Щоденний прийом фолієвої кислоти до- та в перший місяць вагітності суттєво знижує ризик народження дитини з важкими вродженими вадами головного та спинного мозку (дефектами нервової трубки – ДНТ) і, можливо, з іншими вадами, такими, як розщелини губи та піднебіння.
“March of Dimes” рекомендує всім жінкам дітородного віку щоденно вживати мультивітаміни, до складу яких входить 400 мікрограм фолієвої кислоти та додатково споживати продукти багаті на фолати (природний стан фолієвої кислоти). Це єдина можливість забезпечити організм цією життєво необхідною речовиною. Продукти, багаті на фолієву кислоту (фолати): апельсиновий сік, зелені листові овочі, боби. Важливо знати, що синтетична фолієва кислота (в препаратах) засвоюється організмом набагато краще ніж фолати харчових продуктів. І тому невідомо, чи вживання 400 мікрограм фолатів харчових продуктів щодня може забезпечити такий самий рівень захисту від вроджених вад розвитку як 400 мікрограм синтетичної фолієвої кислоти (тому що, наприклад деякі харчові фолати руйнуються при термічній обробці продуктів).
Жінки, які вже мали дитину з вадами нервової трубки повинні проконсультуватися із своїм лікарем для вибору оптимальної дози фолієвої кислоти. Дослідження показали, що у цих жінок значно знижується ризик народження наступної хворої дитини при застосуванні вищих доз фолієвої кислоти (звичайно рекомендують 4 міліграми щоденно, починаючи прийом за місяць до настання вагітності та протягом першого триместру).
Чи необхідне преконцепційне (перед зачаттям) консультування?
У звіті за 1989 рік комісія експертів рекомендує щонайменше одне преконцепційне консультування для оцінки стану здоров’я жінки.
Візит до лікаря – спеціаліста з питань планування сім’ї особливо важливий для жінок, які вже мали проблеми протягом попередніх вагітностей (народження мертвих або недоношених дітей), для виявлення причин виникнення даних ускладнень та їх усунення, а отже, для планування здорової вагітності. Це консультування необхідне для жінок, які мають хронічні захворювання, такі як цукровий діабет, що підвищує ризик народження дитини з вродженими вадами розвитку.
Під час візиту лікар повинен детально вияснити історію захворювання жінки, результати попередніх вагітностей (якщо такі були), наявність шкідливих факторів оточення.
Які причини, що призводять до проблем вагітності, можна виявити перед зачаттям?
Проведення скринінгу (лабораторного обстеження кожної жінки) на наявність інфекцій:
- Краснуха: жінки, які не хворіли на краснуху в дитинстві, повинні зробити щеплення перед зачаттям дитини. В такому випадку вагітність слід планувати не раніше ніж через 3 місяці після щеплення.
Токсоплазмоз: деякі лікарі рекомендують жінкам обов’язкове обстеження на наявність антитіл до токсоплазми. Ця інфекція у жінки може не викликати ніяких клінічних проявів, однак у плода виникають важкі вади розвитку. Інфікування токсоплазмою можливе при вживанні сирого або недостатньо термічно обробленого м’яса та при контактах з котами. - Гепатит В: Центр по контролю за захворюваннями (США) рекомендує скринінг-обстеження (всім жінкам) на наявність гепатиту В. Неліковані діти інфікованих матерів мають високий ризик захворіти. Преконцепційна вакцинація неінфікованих жінок може запобігти інфікуванню та захворюванню плода.
- Вітряна віспа: жінкам, які не хворіли на вітряну віспу рекомендується щеплення. Планування вагітності не раніше ніж через 3 місяці.
- Інші інфекції: обстеження може виявити інфекційні захворювання сечостатевої системи, які можуть викликати загрозу переривання вагітності в різних термінах.
- Важка ступінь ожиріння також може ускладнювати перебіг вагітності. Дослідження, проведені у 1996 виявили, жо у жінок, які мають ожиріння, ризик народження дитини з ДНТ підвищений у 2-4 рази, порівняно з контрольною групою.
Фахівець з питань планування сім’ї також може вияснити наявність деяких спадкових захворювань. За результатами аналізів крові можна виявити носіїв наступних спадкових захворювань:
- Хвороба Тея-Сакса: важке прогресуюче захворювання, при якому уражається центральна нервова система, лікування немає. Поширене серед євреїв Ашкеназі.
- Серповидно-клітинна анемія: захворювання крові, що уражає переважно чорношкіре населення.
- Таласемія: захворювання крові, поширене в деяких районах Африки, Південної Азії та Середземномор’я.
Виявлення носійства цих захворювань дає змогу спрогнозувати ризик народження хворої дитини (якщо носіями хвороби є обоє батьків). Це також дозволяє вибрати оптимальний варіант ведення вагітності та шляхи пренатальної (допологової) діагностики захворювання у майбутньої дитини.
Хромосомні захворювання, такі як хвороба Дауна, мають чітку залежність від віку жінки, тобто, чим старша жінка, тим більший ризик народження хворої дитини. У випадках, коли вагітність виношують жінки, віком 30 років і старші, генетичне консультування необхідне.
Які ще хвороби матері можуть становити небезпеку для майбутньої дитини?
- Цукровий діабет: жінки з недостатньо контрольованим інсулін-залежним діабетом мають підвищений ризик народження дитини з важкими вродженими вадами розвитку а також мертвонароджень та невиношування вагітності. Контроль та корекція вмісту глюкози в крові перед та протягом вагітності підвищує шанси на народження здорової дитини.
- Високий артеріальний тиск: хронічна гіпертонія може стати причиною наступних ускладнень під час вагітності: затримки росту плода та плацентарних проблем. Лікуючий лікар повинен знати про наявність у жінки гіпертонії перед вагітністю для того щоб вибрати правильну тактику її ведення та лікування.
- Системний червоний вовчак: аутоімунне захворювання, яке клінічно проявляється артритами, ураженням нирок та висипкою на шкірі та ін., може стати причиною мертвонародження та невиношування вагітності. Однак, якщо хвора жінка завагітніла після того, як симтоми захворювання не проявлялися протягом останніх 6-ти місяців, вона може мати здорову вагітність.
- Судоми: деякі протисудомні препарати, які приймає жінка під час вагітності підвищують ризик виникнення вроджених вад у плода.
- Фенілкетонурія (ФКУ): жінки з цим спадковим дефектом обміну амінокислот повинні дотримуватися спеціальної дієти до- та протягом усієї вагітності, щоб попередити народження дитини з розумовою відсталістю та вродженими вадами. Лікар повинен проводити постійний контроль за рівнем фенілаланіну в крові такої вагітної та відповідно коригувати дієту.
- Препарати: під час візиту до лікаря кожна жінка може вияснити наскільки безпечні ті чи інші препарати, які вона приймає, для майбутньої вагітності. Це відноситься і до препаратів, які вживають рідко (наприклад при болях голови та при простуді).
Чи може спосіб життя зашкодити майбутній дитині?
Жінки, які зловживають алкоголем мають високий ризик народження дитини з так званим фетальним алкогольним синдромом, який включає фізичні та психічні вади.
Паління подвоює ризик розвитку позаматкової вагітності та народження гіпотрофічної (з низькою вагою) дитини. Вживання кокаїну в ранніх термінах вагітності підвищує ризик народження дитини з вадами розвитку та невиношування вагітності. У жінок, що продовжують вживати наркотики, ризик народження дітей з низькою вагою збільшується в 6 разів.
Користування гарячою ванною чи сауною в ранніх термінах вагітності може стати причиною виникнення у плода вроджених вад розвитку нервової трубки.
Вживання 1-1/2 чашок кави щоденно може знизити можливість завагітніти та підвищує ризик невиношування вагітності.
Спосіб життя звичайно можна змінити заради здоров’я майбутньої дитини, але багато вагітностей є незапланованими. Отже, коли виявляється незапланована вагітність, важливо покинути шкідливі звички та якнайшвидше відвідати лікаря.
Чи потребують преконцепційної консультації чоловіки?
Такі фактори, як шкідливості на виробництві, паління та вживання алкоголю чоловіками відіграють роль у перебігу вагітності. Оскільки ці впливи ще недостатньо вивчені, чоловіки в будь-якому випадку повинні покидати шкідливі звички перед зачаттям та отримати консультацію фахівця про вплив шкідливих факторів на виробництві на здоров’я майбутньої дитини.
Переглянуто редакційною колегією I.B.I.S.: 05/02/2002
|
|
|
|
|
|




