
Serhiy
Синдром Рубінштейна-Тейбі
(Rubinstein–Taybi Syndrome)
Ірина Сергіївна Кулик
Студентка Хмельницького державного університету
Що таке синдром Синдром Рубінштейна-Тейбі (Р-Т)?
Синдром Р-Т – це специфічний розлад, при якому певні фізичні ознаки та аномалії розвитку виникають разом. Особи із цим синдромом зазвичай невисокі на зріст, відстають у розвитку, мають характерні риси обличчя та широкі перші пальці рук та ніг. Вперше синдром був описаний у 1963 році Джеком Рубінштейном та Хоошангом Тейбі (Jack Rubinshtein, Hooshang Taybi), які помітили такі ознаки у семи пацієнтів. З 1963 року було діагностовано понад 400 випадків синдрому Р-Т. Вважається, що цей синдром зустрічається з частотою 1 на 30000 народжень. Однаково часто хворіють дівчатка та хлопчики.
Як розпізнати синдром Р-Т?
Більшість дітей із синдромом Р-Т при народженні не схожі на своїх батьків. Встановити синдром при народженні дуже важко (середній вік встановлення діагнозу – 15 місяців), іноді він не проявляється протягом багатьох років. На жаль, не існує медичних тестів, щоб допомогти встановити синдром Р-Т. Єдиний можливий спосіб – помітити специфічні риси. Характерними рисами обличчя є – маленька голова, низький ріст волосся (волосся жорстке та іноді росте низько на лобі), антимонголоїдний розріз очей, гострий ніс, маленький рот та високе піднебіння. Специфічними є також широкі великі пальці на руках та ногах. Більшість цих ознак важко розпізнати настільки, щоб це було підставою для встановлення діагнозу. Ці риси можуть бути непомітними для родичів та друзів, але дозволять спеціалісту-генетику або педіатру діагностувати синдром Р-Т.
Що є причиною синдрому Р-Т?
Причини виникнення синдрому Р-Т невідомі. При цьому синдромі не встановлено ніяких аномалій хромосом. Однак, не виключається мутація гена. Не підтверджено також вплив хімічних чи інших речовин під час вагітності, що можуть бути причиною виникнення синдрому. Генетична природа синдрому Р-Т не підтверджена.
Чи є синдром Р-Т спадковим?
У більшості випадків, синдром зустрічається лише у однієї дитини певної родини. У медичній літературі згадувалось про сім сімейних випадків синдрому Р-Т, однак більшість з них були описані недостатньо точно. Серед них згадувались два ймовірні випадки синдрому Р-Т, де брат та сестра у одній родині мали цей синдром, а у іншій – синдром діагностували у матері та сина. З огляду на це, ризик народження ще однієї дитини із синдромом у родині становить близько 1 відсотка, тоді як ризик народження дитини особою, якій діагностували цей синдром, складає 50 відсотків. Методи пренатальної діагностики синдрому Р-Т не розроблені.
Що можна очікувати у перші місяці після народження дитини із синдромом Р-Т?
Більшість дітей із синдромом Р-Т народжуються недоношеними. Однак, ускладненням майже третини вагітностей плодом із синдромом Р-Т є багатовіддя. Приблизно кожна п’ята дитина із цим синдромом потребуватиме інтенсивної терапії в періоді новонародженості. Можуть виникнути такі проблеми як затруднене дихання, проблеми із вигодовуванням, повільний темп набирання маси, схильність до закрепів.
Які інші медичні труднощі можуть виникнути?
У дітей з синдромом Р-Т можуть спостерігатися різні проблеми із здоров’ям. Однак, зрозуміло, що в окремої дитини не обов’язково зустрінуться усі перераховані нижче ознаки. Більшість дітей у перші місяці життя мають малу масу тіла, що викликано проблемами із вигодовуванням, такими як поганий апетит, блювання та утруднене ковтання. У деяких випадках госпіталізація – єдина умова нормального лікування. Однак, проблеми із харчуванням вирішуються протягом першого року життя.
- Проблеми із зубами зустрічаються у 2/3 усіх дітей із синдромом Р-Т. Як правило, це порушення розташування зубів та збільшення проміжків між ними. Характерними для осіб із синдромом Р-Т є високе піднебіння, що зумовлює необхідність консультації ортодонта для більшості пацієнтів. Часто спостерігаються гострі відростки на зворотній стороні постійних різців із своєрідною структурою, вони можуть містити пульпу. У деяких випадках вони можуть спричинити збільшення відстані між зубами або подразнення язика, що часом потребує тривалого лікування.
- Більше 80% дітей із синдромом Р-Т мають різноманітні вади очей, включаючи страбізм (косоокість), катаракти, стеноз носослізного каналу. Вчасно не діагностована глаукома, що є при народженні або з’являється у перші місяці життя, може призвести до сліпоти. Саме через це, вкрай необхідно, щоб кожна дитина була обстежена офтальмологом. Часті інфекції вух спостерігаються у половини пацієнтів із синдромом Р-Т, у чверті осіб із цим синдромом – помірна втрата слуху. Повторні респіраторні захворювання також є звичними у перші роки життя та у дитинстві.
- Вроджені вади серця спостерігаються у 35-40% пацієнтів із синдромом Р-Т. Деякі з них є незначними, тоді як інші – складні і вимагають хірургічного лікування.
- Більшість хлопчиків із синдромом Р-Т мають неопущені яєчка, тоді хірургічна корекція є необхідною.
- Ще однією складною проблемою для пацієнтів із цим синдромом є закрепи. Переважна більшість дітей потребують спеціальної дієти, клізми, свічки, та найкраще про це проконсультуватися із педіатром.
- Розширені фаланги перших пальців спостерігаються в усіх пацієнтів із синдромом Р-Т. Інші пальці також можуть бути трохи ширшими, ніж звичайно. Якщо великі пальці ще й кутовидні, то необхідна хірургічна корекція для відновлення їх функцій. У таких випадках слід звернутися до хірурга, що спеціалізується на таких операціях. Дуже широкі великі пальці ніг можуть потребувати хірургічної корекції для зручності носити взуття. Інші можливі ортопедичні труднощі включають зміщення колінних чашечок та сколіоз (викривлення хребта). Лікар, оглядаючи дитину, повідомить про наявність чи відсутність останніх.
- У літературних джерелах повідомляється про схильність до утворення рубців у пацієнтів із синдромом Р-Т, особливо рубці можуть утворюватись після хірургічних операцій, травм і навіть після перенесеної вітрянки.
- Може зрости ризик виникнення аритмії при анестезії. Хоча це вимагає подальшого більш детального дослідження, батьки та анестезіологи повинні це враховувати.
Не в усіх дітей із цим синдромом зустрічаються усі перераховані тут проблеми, деякі діти мають складні хронічні захворювання, тоді як у інших спостерігаються незначні відхилення.
Чи потрібно дітям із синдромом Р-Т робити щеплення?
Загальний рівень ускладнень, які проявлялись, в основному, незначним підвищенням температури та роздратованістю, склав лише 5%, що не набагато більше ніж у здоровій популяції. Тому немає необхідності уникати щеплення, які можуть попередити серйозні інфекційні хвороби.
Ріст та розвиток дитини із синдромом Р-Т
Більшість дітей із синдромом Р-Т при народженні мають середні значення зросту та ваги. Однак, в перші місяці життя стає очевидною затримка росто-вагових показників, відсутні ростові “стрибки” у підлітковому періоді. Кінцевий зріст чоловіків становить 152 см, жінок – 125 см, проте можливі міжсімейні варіації. Хлопчики, зазвичай, схильні до надмірної ваги у шкільні роки, тоді як у дівчаток надмірна вага спостерігається у юності.
Статева зрілість та здатність народжувати дітей
Згідно спостережень, статеве дозрівання починається у 12 років, як і в здоровій популяції. Менструальний цикл у дівчаток так само починається у звичний період. В медичних довідниках згадуються два випадки, коли жінки із синдромом Р-Т народжували дітей. В однієї був син із цим синдромом та здорова донька. Інша також мала здорову дитину. Слід зазначити, що особи із синдромом Р-Т здатні мати дітей.
Розвиток
Усі діти із синдромом Р-Т розумово відсталі. Однак, ступінь відсталості відрізняється у кожному окремому випадку. Коефіцієнт інтелекту (IQ) у осіб із синдромом Р-Т коливається в межах 30-79 (нормальний рівень – 85-115). Зазвичай, особи із цим синдромом мають так званий помірний рівень розумової відсталості. Деякі пацієнти демонструють глибоку розумову відсталість, тоді як інші досягають нижньої межі нормального розвитку. Найвищий рівень інтелекту у дитини із синдромом Р-Т, зазначений у літературі, складає 80. Відзначається також, що дитина, яка виховується у сім’ї, має значно вищий рівень інтелекту, ніж та, яка зростає в інтернаті. Однак, важко передбачити успіхи дитини із синдромом Р-Т, оскільки кожний окремий випадок є унікальним.
90% пацієнтів із синдромом Р-Т мають проблеми із мовленням. Деякі також мають проблеми із артикуляцією. Невелика кількість дітей використовує для мовлення лише жести, тоді як майже половина – і слова і жести разом. Переважна більшість дітей потребують мовної терапії.
Діти із синдромом Р-Т розвиваються і отримують нові знання протягом усього життя. Більшість дітей починають готуватися до занять ще в ранньому дитинстві. Вони проходять спеціальну терапію та навчання. Для дітей із фізичними вадами можливо потрібна буде спеціальна програма підготовки. Старші особи із синдромом Р-Т проходять спеціальні програми тренування для виконання нескладних операцій. Деякі особи можуть жити майже самостійно, тоді як інші назавжди залишаються жити із своїми родинами.
Кожна дитина із синдромом Р-Т є унікальною особистістю. Живучи в родині, яка любить і поважає її, дитина розвивається і є щасливою. Діти цікавляться музикою, спортом, книгами та телебаченням.
Поведінка
Іноді батьки стурбовані поведінкою їхньої дитини із синдромом Р-Т: деякі діти можуть крутитись на місці, перекочуватись, плескати в долоні, що є проявом самостимулюючої поведінки. Слабка концентрація уваги відзначається у 90% пацієнтів, і лише незначна кількість дітей є занадто активними. Усі діти по різному реагують на звук: декого дратують голосні звуки, дехто не любить натовп через шум. Лікар або вихователь може порадити консультацію спеціаліста, щоб вирішити проблему із поведінкою хворої дитини.
Переглянуто редакційною колегією I.B.I.S.: 20/01/2004
Дивіться також:
Синдром ламкої Х-хромосоми – об’єм яєчок
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”.
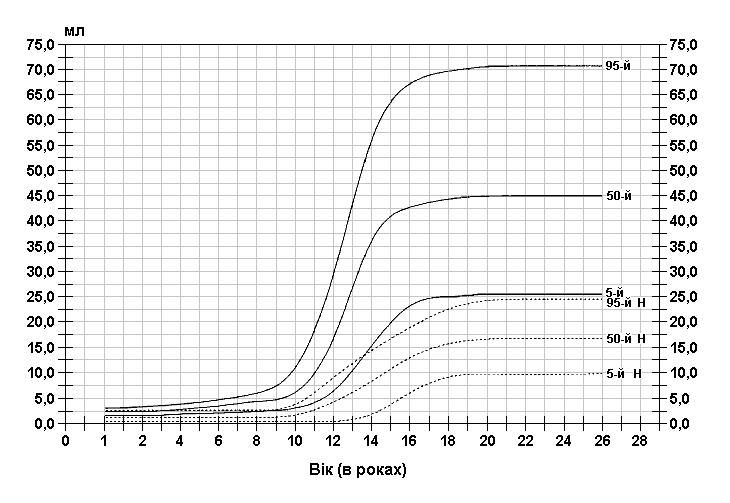
Стандартизовані криві збільшення об’єму яєчок у хлопчиків з синдромом ламкої Х-хромосоми (суцільна лінія) і у здорових дітей (пунктирна лінія). Дані отримані в результаті обстеження 185 хлопчиків від народження до 26-річного віку, які відносяться до білошкірої раси та мають синдром ламкої Х-хромосоми. Діагноз підтверджувався результатами цитогенетичних досліджень (діапазон експресії від 4 до 58%, середній – 21%). Butler MG, Brunschwig A, Miller LK, Hagerman RJ: Pediatrics 89:1059-1062, 1992.
Синдром ламкої Х-хромосоми – довжина вуха у хлопчиків
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”.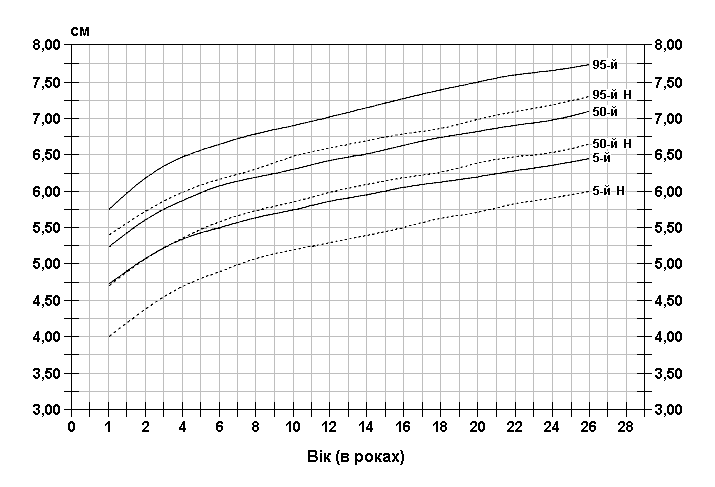
Стандартизовані криві збільшення довжини вуха у хлопчиків з синдромом ламкої Х-хромосоми (суцільна лінія) і у здорових дітей (пунктирна лінія). Дані отримані в результаті обстеження 185 хлопчиків від народження до 26-річного віку, які відносяться до білошкірої раси та мають синдром ламкої Х-хромосоми. Діагноз підтверджувався результатами цитогенетичних досліджень (діапазон експресії від 4 до 58%, середній – 21%). Butler MG, Brunschwig A, Miller LK, Hagerman RJ: Pediatrics 89:1059-1062, 1992.
Завантажити діаграму в форматі pdf (Acrobat Reader, 119 кб)
Синдром ламкої Х-хромосоми – обвід голови у хлопчиків
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”.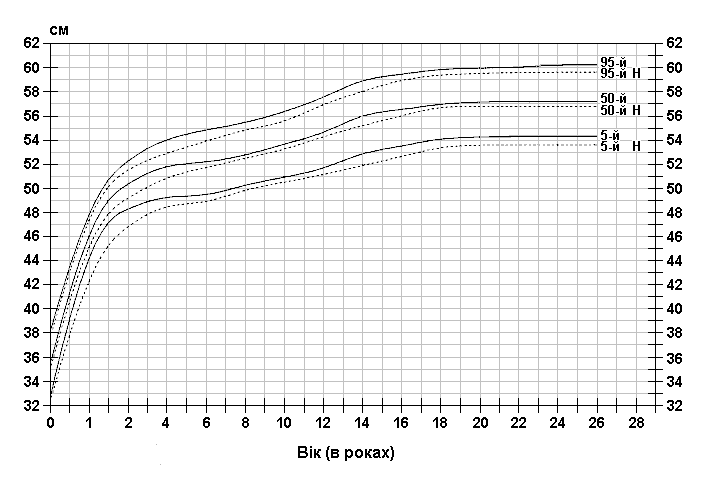
Стандартизовані криві збільшення обводу голови у хлопчиків з синдромом ламкої Х-хромосоми (суцільна лінія) і у здорових дітей (пунктирна лінія). Дані отримані в результаті обстеження 185 хлопчиків від народження до 26-річного віку, які відносяться до білошкірої раси та мають синдром ламкої Х-хромосоми. Діагноз підтверджувався результатами цитогенетичних досліджень (діапазон експресії від 4 до 58%, середній – 21%). Butler MG, Brunschwig A, Miller LK, Hagerman RJ: Pediatrics 89:1059-1062, 1992.
Завантажити діаграму в форматі pdf (Acrobat Reader, 129 кб)
Синдром ламкої Х-хромосоми – зріст хлопчиків
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”.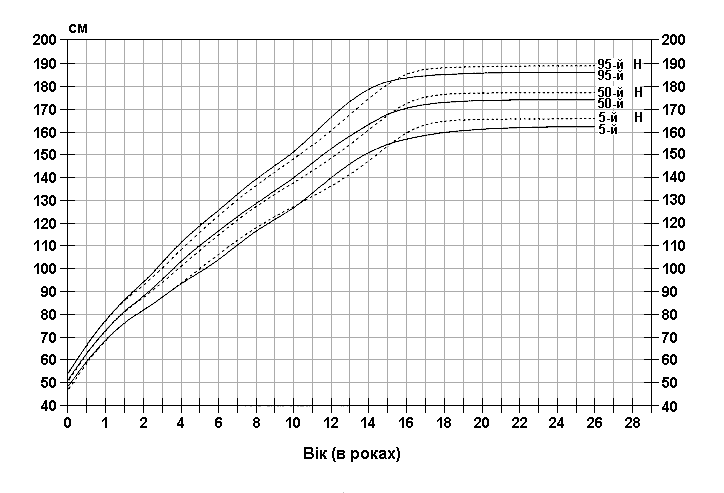
Стандартизовані криві збільшення росту у хлопчиків з синдромом ламкої Х-хромосоми (суцільна лінія) і у здорових дітей (пунктирна лінія). Дані отримані в результаті обстеження 185 хлопчиків від народження до 26-річного віку, які відносяться до білошкірої раси та мають синдром ламкої Х-хромосоми. Діагноз підтверджувався результатами цитогенетичних досліджень (діапазон експресії від 4 до 58%, середній – 21%). Butler MG, Brunschwig A, Miller LK, Hagerman RJ: Pediatrics 89:1059-1062, 1992.
Завантажити діаграму в форматі pdf (Acrobat Reader, 118 кб)
Синдром ламкої Х-хромосоми – вага хлопчиків
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”.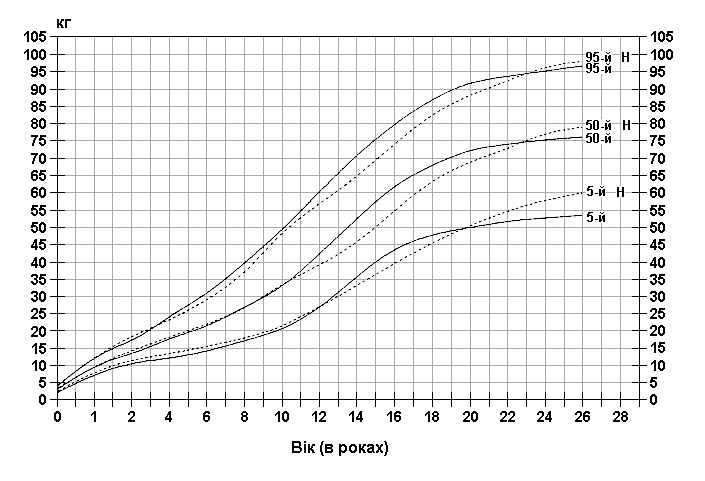
Стандартизовані криві збільшення ваги у хлопчиків з синдромом ламкої Х-хромосоми (суцільна лінія) і у здорових дітей (пунктирна лінія). Дані отримані в результаті обстеження 185 хлопчиків від народження до 26-річного віку, які відносяться до білошкірої раси та мають синдром ламкої Х-хромосоми. Діагноз підтверджувався результатами цитогенетичних досліджень (діапазон експресії від 4 до 58%, середній – 21%). Butler MG, Brunschwig A, Miller LK, Hagerman RJ: Pediatrics 89:1059-1062, 1992. Copyright © 1992 Reproduced by permission of Pediatrics.
Завантажити діаграму в форматі pdf (Acrobat Reader, 118 кб)
TAR-синдром
(TAR Syndrome)
Інна Тарасівна Мороз
Лікар-неонатолог відділення патології новонароджених
Рівненської обласної дитячої лікарні
Включення:
- Тромбоцитопенія з відсутністю променевої кістки.
- Синдром тетрафокомелії-тромбоцитопенії.
Основні діагностичні критерії:
Тромбоцитопенія, двобічна відсутність або гіпоплазія променевих кісток.
Клінічна характеристика:
Тромбоцитопенія спостерігається у 100% випадків і з’являється в перші 4 місяці життя; з віком кількість тромбоцитів підвищується до нормального рівня. Стресові ситуації, інфекції та хірургічне втручання провокують тромбоцитопенічні кризи. Порушується агрегація тромбоцитів, відповідно скорочується час їхнього життя. Мегакаріоцити можуть бути функціонально неактивними (12%) або бути відсутніми (66%); зменшення кількості і розмірів мегакаріоцитів спостерігається у 12% випадків. У 60-70% випадків на першому році життя, особливо в період кровотеч, відмічається лейкемоїдна реакція; в цей час наростає тромбоцитопенія і з’являється гепатоспленомегалія. Зустрічається еозинофілія, анемія. Мелена, гематемезис.
Основний дефект скелету – двобічна відсутність або гіпоплазія променевої кістки (100%), часто в поєднанні з аномаліями кисті (обмеження розгинання, радіальна девіація, гіпоплазія карпальних кісток і фаланг з обов’язковим збереженням І пальця); гіпоплазією (100%), двобічною відсутністю (20%) і/або однобічною відсутністю (10%) ліктьової кістки, аномаліями плеча (50%), з двобічною його відсутністю у 5-10%.
У дорослих хворих немає інших симптомів, окрім менорагії. Внутрішньочерепні крововиливи зазвичай спостерігаються у віці до 1 року. Затримка моторного розвитку внаслідок скелетних деформацій. Пізнім ускладненням захворювання є артрит суглобів кистей та колінних суглобів. Проявом шкірних змін є пурпура. Носові кровотечі.
Поєднані аномалії:
Іноді зустрічаються вивихи кульшових суглобів та надколінника, підвивих колінних суглобів, тугорухливість колінного суглоба, вальгусна деформація нижніх кінцівок, клишоногість, тетрафокомелія, аномалії ребер та хребта, в тому числі спинномозкова кила, брахіцефалія, синдактилія, вроджені вади серця (30%), найчастіше тетрада Фалло і дефект міжпередсердної перегородки; косоокість, глаукома. Інколи зустрічаються ниркова патологія, дивертикул Меккеля. Можлива розумова відсталість (7%), пов’язана з внутрішньочерепними крововиливами, низький зріст, набряк тилу ступнів, надмірне потовиділення, мікрогнатія, кисти підшлункової залози, гіпогамаглобулінемія, гіпоплазія мозочка, часткова – хробака. Гемангіома і судинний невус. Гіпоплазія/гіпогенезія нижньої, верхньої щелепи. Зустрічалися випадки із розщілиною м’якого піднебіння, стенозом гортані, дуоденальна атрезія.
Диференційний діагноз:
Синдром панцитопенії Фанконі, синдром Холта-Орама, синдром Робертса.
Прогноз:
Біля 40% пацієнтів помирають, зазвичай внаслідок кровотеч в ранньому дитинстві.
Пренатальний діагноз:
Огляд верхніх кінцівок під час проведення сонографії.
Тип успадкування: аутосомно-рецесивний.
Співвідношення статей: Ч1 : Ж1.
Номер з каталогу МІМ:
274000 Thrombocytopenia-Absent Radius Syndrome; TAR.
Література:
- Наследственные синдромы и медико–генетическое консультирование /С.И. Козлова, Н.С. Демикова, Е. Семанова, О.Е. Блинникова.- М.: Практика, 1996.- С. 268-270.
- Jones KL. Smith’s Recognizable Patterns of Human Malformation. W.B.Saunders Company, Philadelphia 1997:322-323.
- Thrombocytopenia-Absent Radius Syndrome OMIM #274000.
- Warkany J. Congenital Malformations. Notes and Comments. Year Book Publisher. Chicago 1981.
Переглянуто редакційною колегією I.B.I.S.: 05/01/2004
Синдром Холта-Орама
(Holt-Oram Syndrome)
Ярослава Анатоліївна Нестеренко
Лікар акушер-гінеколог-генетик
Ніжинського пологового будинку
Синоніми: синдром руки-серця тип І.
Популяційна частота: 1:100000.
Етіологія:
Аутосомно–домінантний синдром із майже 100% пенетрантністю. Розвиток синдрому обумовлений мутацією гену TBX5, який картовано у сегменті q2 хромосоми 12.
Діагностичні ознаки:
Вроджені вади розвитку верхньої кінцівки, вроджені вади серця.
Клініка:
- Вади розвитку верхньої кінцівки (частіше лівої), різного ступеня вираженості – від фокомелії до малих аномалій, що виявляються тільки рентгенологічно (гіпоплазія та аплазія малих кісток зап’ястя, іноді їх злиття між собою).
Найбільш типовим є ураження І пальця китиці у вигляді дігіталізації, трьохфалангового І пальця, гіпоплазії та аплазії його. У третини хворих спостерігаються дефекти інших пальців – клинодактилія, синдактилія, гіпоплазія та ін. Близько половини пацієнтів мають гіпоплазію та аплазію променевої кістки. В деяких випадках спостерігається гіпоплазія ліктьової кістки. Дефекти кінцівок можуть супроводжуватися іншими аномаліями кістяка (20-25% випадків): аномаліями плечових кісток, лопаток (поворот в сторону аксілярних ліній), ключиць (розширення зовнішнього кінця голівки), відсутність великого грудного м’яза, гіпоплазія ребер, лійкоподібна грудна клітка, сколіоз. - Вроджені вади серця виявляються у понад 85% хворих. Переважно це дефект міжпередсердної перетинки, рідше дефекти міжшлуночкової перетинки, відкрита артеріальна протока, коарктація аорти, стеноз легеневої артерії, пролапс мітрального клапана.
Вади розвитку інших органів при синдромі Холта-Орама не характерні. Розумовий розвиток пацієнтів не страждає. Синдром Холта Орама може бути виключений у пацієнтів які мають тільки вади розвитку ліктьової кістки, вади розвитку нирок, хребців, нижніх кінцівок, очей, ануса, черепно–лицьові аномалії, порушення слуху або аномалії вуха.
Диференційний діагноз:
Діагностика синдрому в сімейних випадках не становить труднощів. У спорадичних випадках (нові мутації) необхідно диференцівати з:
- панцитопенією Фанконі (ОМІМ 227650);
- VATER-ассоціацією (ОМІМ 192350);
- TAR-синдромом (ОМІМ 274000);
- синдромом Едвардса (ОМІМ 300484);
- синдромом Орбелі;
- хондроектодермальною дисплазією Елліса-ван-Кревельда (ОМІМ 225500);
- фетальним алкогольним синдромом.
Співвідношення статей: Ч1 : Ж1.
Тип успадкування:
Аутосомно-домінантний із практично 100% пенетрантністю.
Вік маніфестації: з народження.
Лікування:
Хірургічна корекція вроджених вад розвитку.
Прогноз:
Залежить від типу вродженої вади серця. При її корекції прогноз для життя сприятливий.
Медико-генетичне консультування:
Ризик для нащадків – 50%, ризик для сибсів при нових мутаціях не перевищує загально-популяційного.
Пренатальна діагностика:
Ґрунтується на виявленні у плоду поєднаних вад розвитку верхніх кінцівок та вад серця. Для диференціації з синдромами Едвардса та Орбелі проводиться інвазивна пренатальна діагностика з визначенням каріотипу плода.
Номер з каталогу МІМ:
142900 Holt-Oram Syndrome; HOS.
Література:
- Козлова С.И. Наследственные синдромы и медико-генетическое консультирование: Атлас–справочник.- Изд. 2-е дополн. – М.: Практика, 1996.- 416 с.
- Basson CT, Cowley GS, Solomon SD et al. The clinical and genetic spectrum of the Holt-Oram syndrome (heart-hand syndrome) N Engl J Med 1994;330:885–891.
- Buyse ML. Birth defects encyclopedia. Center for Birth Defects Information Services, Inc. 1990.
- Cross SJ, Ching YH, Li QY et al. The mutation spectrum in Holt-Oram syndrome. J Med Genet 2000;37:785–787.
- Holt M, Oram S. Familial heart disease with skeletal malformations. Br Heart J 1960;22:236–242.
- Huang T. Current advances in Holt-Oram syndrome. Curr Opin Pediatr 2002;14:691–695.
- Newbury-Ecob RA, Leanage R, Raeburn JA et al. Holt-Oram syndrome: a clinical genetic study. J Med Genet 1996;33:300–307.
- Sletten LJ, Pierpont ME. Variation in severity of cardiac disease in Holt-Oram syndrome. Am J Med Genet 1996;65:128–132.
Переглянуто редакційною колегією I.B.I.S.: 26/05/2005
Синдром “Femur-fibula-ulna” (FFU)
(Femur-Fibula-Ulna Syndrome)
Оксана Миколаївна Масняк
Лікар-генетик Рівненського обласного клінічного
лікувально-діагностичного центру ім. В. Поліщука
Синоніми:
- Дизостоз “Femur-fibula-ulna”;
- Proximal focal femoral deficiency (PFFD).
Основні діагностичні критерії:
Асиметричне вкорочення малогомілкової, стегнової і ліктьової кісток.
Ознаки:
Вкорочення малогомілкової, стегнової, ліктьової кісток, а іноді навіть повна відсутність кінцівок. Вкорочення стегнових кісток створює враження надлишку шкіри на стегнах. При відсутності верхніх кінцівок характерною ознакою синдрому є втиснення шкіри, яке схоже з післяампутаційним; лопатка і ключиця розвинуті нормально. Нерідко спостерігається синостоз плечової і променевої кісток, дефекти розвитку латеральних п’ясних кісток і відповідних їм фаланг. Дефекти кінцівок у більшості випадків асиметричні. Верхні кінцівки уражаються частіше, ніж нижні; права частина тіла уражається частіше, ніж ліва. Психомоторний розвиток відповідає віку. Аномалії внутрішніх органів зустрічаються рідко.
Класифікація:
Виділяють 4 форми:
- І форма – ураження однієї кінцівки;
- ІІ форма – ураження двох кінцівок;
- ІІІ форма – ураження трьох кінцівок;
- ІV форма – ураження усіх чотирьох кінцівок.
Найчастіше зустрічається І форма.
Дані обстежень:
Рентгенологічне обстеження виявляє ураження кісток кінцівок. Можлива пренатальна діагностика.
Диференційний діагноз:
АДАМ-комплекс, синдром Холта-Орама, синдром тромбоцитопенії з відсутністю променевої кістки, синдром Корнелії де Ланге, ЕЕС синдром, анемія Фанконі, синдром гіпоплазії стегна і незвичайного обличчя, синдром Робертса, рото-лице-пальцевий синдром ІІ тип, синдром Рассела-Сільвера.
Етіологія:
Всі випадки спорадичні. Тип успадкування невідомий. Деякі автори висувають теорію впливу ранньої соматичної мутації, як причину синдрому, але ці дані не доведені.
Частота виникнення:
0,11-0,2 на 10000 новонароджених.
Ризик для сибсів: низький.
Ризик для нащадків: низький.
Співвідношення статей:
Ч:Ж = 1:1, за іншими даними, чоловіча стать уражається частіше, ніж жіноча [1].
Вік прояву: з народження.
Прогноз:
Рання інвалідизація. Ступінь порушення функцій самообслуговування залежить від форми захворювання.
Профілактика:
Пренатальна діагностика і медико-генетичне консультування.
Лікування:
Ортопедичне лікування залежить від виду дефекту.
Номер з каталогу МІМ:
228200 Femur-Fibula-Ulna Syndrome.
Література:
- Аномалии развития органов и частей тела человека: Справ. пособие /О.В. Калмин, О.А. Калмина. – Пенза: Изд-во Пенз. гос. ун-та, 2004. – С. 246-247.
- Козлова С.И., Демикова Н.С., Семанова Е., Блинникова О.Е. Наследственные синдромы и медико-генетическое консультирование.- М.: Практика, 1996.- C. 255.
- Benacerraf BR. Ultrasound of fetal syndromes. 1st ed. 1998:145-146,364-365.
- Human Malformations and Related Anomalies. Volume II. Edited by Stevenson RE, Hall JG, Goodman RM. Oxford Univ. Press 1993:703-718.
Переглянуто редакційною колегією I.B.I.S.: 22/02/2011
CHILD Синдром
(CHILD Syndrome)
О.П. Семененко
Завідувач Черкаським обласним медико-генетичним центром
Синоніми:
- Ichthyosiform erythroderma, unilateral, with ipsilateral malformations, especially absence deformity of limbs;
- Congenital hemidysplasia with ichthyosiform erythroderma and limb defects.
Назва походить від акронімів аномалій, що входять до складу синдрому: вроджена гемідисплазія (Congenital Hemidisplasia), іхтіозіформна еритродермія (Ichthyosiform Erythroderma), аномалії кінцівок (Limb Defects).
Частота:
Відноситься до рідкісних синдромів, описано близько 30 випадків (29 випадків у хлопчиків та 1 у дівчинки).
Основні діагностичні критерії:
- Вроджена гемідисплазія;
- Іхтіозіформна еритродермія;
- Аномалії кінцівок;
- Однобічні аномалії внутрішніх органів.
Клінічна характеристика:
Шкіра:
- Однобічна еритродермія та лущення з народження або відразу після;
- Чітка лінія демаркації по середній лінії тіла;
- Гіперкератоз.
Кінцівки:
- Однобічна гіпомелія (від гіпоплазії пальців до повної відсутності кінцівки);
- Ліктьовий птеригіум;
- Колінний птеригіум;
- Контрактури суглобів.
Іпсілатеральні аномалії внутрішніх органів:
- Гіпоплазія легенів;
- Гіпоплазія яйників, гіпоплазія фалопієвих труб;
- Агенезія нирок, гідронефроз;
- Гіпоплазія мозку, гіпоплазія черепних нервів;
- Гіпоплазія щитоподібної залози, гіпоплазія наднирників.
Розвиток:
Помірна затримка внутрішньоутробного розвитку. Правий бік уражається частіше лівого, при лівобічному варіанті синдрому спостерігаються важкі вади серця.
Поєднані симптоми:
Туговухість. Щілина верхньої губи. Септальні дефекти серця, єдиний шлуночок, єдине коронарне вустя. Однобічна гіпоплазія ключиці, лопатки, ребер. Пупкова кила. Іпсілатеральна гіпоплазія нижньої щелепи. Сколіоз. Однобічна гіпоплазія кісток тазу. Ламкість нігтів. Однобічна алопеція. Збільшення концентрації 8-дегідрохолестиролу та 8(9) холестенолу.
Лікування: кардіохірургічна корекція вад серця.
Профілактика:
Пошук характерних для синдрому вад кінцівок при проведенні ультразвукового дослідження під час вагітності.
Етіологія:
Х-зчеплений домінантний тип успадкування, з летальністю у гемізиготних чоловіків. Ген картований на короткому плечі Х хромосоми – Хq28. Ген NSDHL кодує кофермент, який приймає участь в синтезі холестерина. Високий рівень холестерину відомий як фактор ризику серцево-судинних хвороб, але організм потребує певну кількість холестерину для розвитку та нормального функціонування. До народження, холестерин взаємодіє з сигнальними білками, які контролюють ранній развиток мозку, кінцівок, статевих органів та інших структур. Низький рівень холестерину і / або накопиченя інших речовин, порушує ріст і розвиток багатьох частин тіла. Це достовірно невідомо, однак порушення синтезу холестерину призводить до розвитку фенотипових особливостей синдрому CHILD.
Номер з каталогу МІМ:
308050 Congenital Hemidysplasia with Ichthyosiform Erythroderma and Limb Defects.
Література:
- Белозеров Ю.М. Детская кардиология. Наследственные синдромы.- 2008.- c.314-315.
- Cullen SI, Harris DE, Carter CH, Reed WB. Congenital unilateral ichthyosiform erythroderma. Arch Derm 1969;99:724-729.
- Emami S, Rizzo WB, Hanley KP, Taylor JM, Goldyne ME, Williams ML. Peroxisomal abnormality in fibroblasts from involved skin of CHILD syndrome: case study and review of peroxisomal disorders in relation to skin disease. Arch Derm 1992;128:1213-1222.
- Falek A, Heath CW Jr, Ebbin AJ, McLean WR. Unilateral limb and skin deformities with congenital heart disease in two siblings: a lethal syndrome. J Pediat 1968;73:910-913.
- Goldyne ME, Williams ML. CHILD Syndrome. Phenotypic dichotomy in eicosanoid metabolism and proliferative rates among cultured dermal fibroblasts. J Clin Invest 1989 Jul;84(1):357-60.
- Happle R, Effendy I, Megahed M, Orlow SJ, Kuster W. CHILD syndrome in a boy. Am J Med Genet 1996;62:192-194.
- Hebert AA, Esterly NB, Holbrook KA, Hall JC. The CHILD syndrome: histologic and ultrastructural studies. Arch Derm 1987;123:503-509.
- Hummel M, Cunningham D, Mullett CJ, Kelley RI, Herman GE. Left-sided CHILD syndrome caused by a nonsense mutation in the NSDHL gene. Am J Med Genet 2003;122A:246-251.
- Jones KL. Smith’s Recognizable Patterns of Human Malformations. 5th Edition. 1997:308-309.
- Konig A, Happle R, Bornholdt D, Engel H, Grzeschik K-H. Mutations in the NSDHL gene, encoding a 3-beta-hydroxysteroid dehydrogenase, cause CHILD syndrome. Am J Med Genet 2000;90:339-346.
Переглянуто редакційною колегією I.B.I.S.: 29/09/2012
|
|
|
|
|
|




