
Serhiy
Статевий розвиток дівчат
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”
Послідовність статевого розвитку –
середньостатистична американська жінка
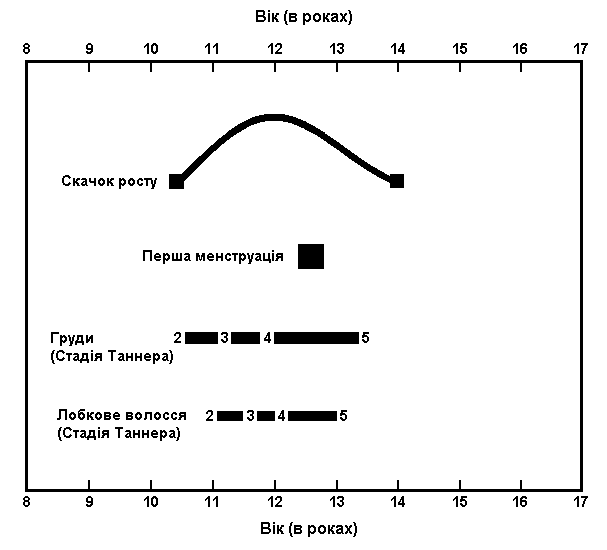
Адаптовано з Brookman et al: The Princeton maturation study, 1976 (використовується з дозволу, Ross Laboratories).
Мультикістоз нирок
(Multicystic Kidneys)
О.О. Кисельова
Лікар-неонатолог
Херсонської дитячої обласної клінічної лікарні
Включення:
Кістозне захворювання нирок, синдром Поттер II типу, мультикістозна дисплазія нирок, диспластична хвороба нирок.
Визначення:
Мультикістозна хвороба нирок – це вроджена ниркова патологія, яка проявляється кістозним переродженням ниркової паренхіми внаслідок первинного розширення збірних канальців. Процес може бути двобічним, однобічним чи сегментарним.
Основні діагностичні критерії, клінічна картина:
Мультикістозна нирка – це конгломерат різного розміру тонкостінних кіст, між якими практично відсутня ниркова паренхіма. Частіше всього нирка збільшена в об’ємі, маса може досягати кількох сотень грамів. Відсутня диференціація шарів, видно лише кісти, різко зменшена кількість нефронів. Миска часто відсутня чи гіпоплазована, але може бути атрезія, дилатація, подвоєння. Гістологічними маркерами служать примітивні протоки, канальці і клубочки фетального типу. Ниркові кісти являють собою термінальні відділи збірних трубочок, можуть бути оточені сполучною тканиною, гладком’язовими волокнами. При односторонньому процесі на протилежній стороні виявляють гідронефроз. Двобічний процес несумісний з життям. Однобічний процес може бути безсимптомним. Але при збільшеній мультикістозній нирці відмічається великий живіт, пухлиноподібний утвір у підребер’ї, приступоподібні болі у животі, поперековій ділянці. Захворювання часто виявляється при інфікуванні сечовивідних шляхів.
Асоційовані вади:
Мультикістоз нирок може бути проявом аутосомно-рецесивних синдромів (Меккеля, Зельвегера, Денді-Уокера, Івемарка, Салдіно-Нунан типу II, Робертса, Фрайнса, Сміта-Лемлі-Опіца, аутосомно-домінантних (синдром Апера), хромосомних порушень.
Дані обстежень:
- Пренатальне ультразвукове дослідження (кістозно змінені нирки збільшених чи зменшених розмірів, недостатня візуалізація сечового міхура плода, маловіддя).
- Ангіографія (виявляється відсутність ниркової ангіограми та нефрограми).
- Внутрішньовенна урографія (виявляється мультикістозно змінена нирка).
- Цистоскопія (часто відсутнє устя сечоводу або наявна облітерація самого сечоводу).
Диференційний діагноз:
- Полікістоз нирок.
- Обструкція на рівні сечовідно-мискового з’єднання.
- Пухлина Вільмса.
- Гематома з вторинним некрозом.
Етіологія:
Пояснюється ембріофетопатією. Захворювання зустрічається в основному спорадично. Іноді хвороба у плода виявлялася при цукровому діабеті у вагітної.
Частота виникнення:
Серед усіх вроджених вад розвитку сечової системи зустрічається у 5,1% випадків, а серед кістозних дисплазій – 25,8%. Процес, як правило, однобічний. Однобічний процес у хлопчиків – 1:500. Двобічний процес – 1:10000.
Патогенез:
Невідомий. Можливо, ця аномалія є результатом двох типів первинних ушкоджень:
- недостатності розвитку метанефрогенної бластеми, відповідальної за формування нефронів;
- ранньої обструктивної уропатії.
Якщо порушення виникають у ранні терміни (8-11 тижнів) гестації, то в нирках визначається 10-20 не з’єднаних між собою кіст, і орган зберігає свою типову конфігурацію. Ниркова миска і чашечки атрезовані чи вкрай малі. Якщо процес розвивається пізніше, то морфологія нирок залежить від тривалості обструкції. Візуалізується типова гідронефротична картина. Пізніше видно збільшення нирки з численними кістами.
Акушерська тактика:
При двобічному процесі, діагностованому у ранніх термінах, слід рекомендувати переривання вагітності. Важливо визначити каріотип плода. Однобічна форма хвороби при нормальному каріотипі і без поєднаних аномалій не впливає на акушерську тактику. У разі виявлення уропатії пологи слід проводити в той момент, коли підтверджена зрілість легенів плода. Пологи рекомендовано проводити у регіональному перинатальному центрі.
Прогноз:
Двобічний процес закінчується смертю хворого. Односторонній мультикістоз нирок – недостатньо даних.
Можливі ускладнення:
Малігнізація, артеріальна гіпертензія.
Лікування:
Симптоматичне і хірургічне. При хронічній нирковій недостатності проводять гемодіаліз, у подальшому показана пересадка нирки.
Номер з каталогу МІМ:
600989 Infundibulopelvic Dysgenesis.
Література:
- Коен А., Наст С. Врожденные аномалии и наследственные заболевания почек //Нефрология.- 1998.- № 4.- C. 90-91.
- Папаян А.В., Савенкова Н.Д. Клиническая нефрология детского возраста.- Санкт – Петербург: Сотис, 1997.- C. 186, 190.
- Bartley JA, Golbbus MS, Filly RA et al. Prenatal diagnosis of dysplastic kidney disease. Clin Genet 1977;11:375.
- Bateman BG, Brenbridge ANAG, Buschi AJ. In utero diagnosis of multicystic kidney disease by sonography. J Reprod Med 1980;25:256.
- Bearman SB, Hine PL, Sanders RC. Multicystic kidney: A sonographic pattern. Radiology 1976;118:685.
- Beretsky I, Lankin DH, Rusoff JH. Sonographic differentation between the multicystic dysplastic kidney and the ureteropelvic junction obstruction in utero using high resolution real-time scanners employing digital detection. J Clin Ultrasond 1984;12:429.
- Bloom DA, Brosman S. The multicystic kidney. J Urol 1978;120:211.
- Chen YH, Stapleton FB, Roy S et al. Neonatal hypertension from a unilateral multicystic, dysplastic kidney. J Urol 1985;133:664.
- Cole BR, Kaufman RL, McAlister WH et al. Bilateral renal dysplasia in three siblings: Report of a survivor. Clin Nephrol 1976;5:83.
- D’Alton M, Romero R, Grannum P et al. Antenatal diagnosis of renal anomalies with ultrasound. IV. Bilateral multicystic kidney disease. Am J Obstet Gynecol 1986;54:532.
- Dunne MG, Johnson ML. The ultrasonic demonstration of fetal abnormalities in utero. J Reprod Med 1979;23:195.
- Felson B, Cussen LJ. The hydronephrotic type of unilateral congenital multicystic disease of the kidney. Semin Roentgenol 1975;10:113.
- Warkany J. Congenital Malformations: Notes and Comments. Chicago: Year Book Publisher, 1981:1042-1048.
Переглянуто редакційною колегією I.B.I.S.: 12/06/2002
Муковісцидоз з погляду сімейного лікаря
(Cystic Fibrosis)
О.М. Мислінчук
Лікар-генетик
Рівненського клінічного лікувально-діагностичного центру
Муковісцидоз (МВ) – це генетичне аутосомно-рецесивне моногенне захворювання, обумовлене мутацією гена трансмембранного регулятора. Для нього характерно порушення секреції екзокринних залоз життєво важливих органів з пошкодженням перш за все дихального та шлунково-кишкового трактів, важким перебігом і несприятливим прогнозом.
МВ вперше виділений із групи целіакій венським педіатром Гвідо Фанконі в 1936 році.
Частота народження хворої дитини серед живонароджених складає 1:2000 – 1:2500, що відносить МВ до найбільш поширених спадкових захворювань. До того ж, 4-5% людей є носіями гена МВ.
Після перших описів МВ вважався фатальним захворюванням, тому що більшість дітей не переживало п’ятирічну межу. До 80-х років 80% хворих не досягало 20-річного віку. Але і в сучасних умовах існує різна тривалість життя хворих, що обумовлено різними рівнями розвитку суспільства, розумінням проблеми, ступенем організації спеціалізованих центрів.
У Великобританії, США, Австралії новонародженим з МВ гарантується 40 років життя, в інших розвинутих країнах – в середньому З1 рік, в Латинській Америці – 10 років (95% хворих тут не виявляється), в Росії за результатами роботи Центру Муковісцидозу на базі республіканської дитячої лікарні (Москва) – 16 років.
Етіологія:
Ген розташований на 7-й хромосомі (7q31). Хвороба має аутосомно-рецесивний тип успадкування з різним ступенем експресивності ознак. Тому ризик повторення народження хворої дитини в сім’ї високий і становить 25%.
В одному гені може бути багато мутацій, кожна з яких характерна для певної популяції або регіону. Тяжкий перебіг характерний для гомозигот по дельта – Р508 (основна мутація, яка передбачає заміну фенілаланіну на 508 місці), а також при мутаціях W / 1282 Х, G б542, N 1303К, як самостійних, так і в поєднанні з дельта – F 508. Відносно легкий перебіг МВ спостерігається у дітей з мутаціями R 334W, S 1196X (іноді в поєднанні з дельта F 508).
Але передбачити перебіг хвороби навіть при одній і тій самій мутації малоймовірно. Іноді навіть при ранній діагностиці та адекватному лікуванні швидко і безповоротно змінюються внутрішні органи. В той же час у деяких пацієнтів при пізній діагностиці і лікуванні спостерігається сприятливий перебіг МВ, який дозволяє досягнути дорослого віку. Такий клінічний поліморфізм можна пояснити генетичними особливостями самої дитини, імунними реакціями, тощо.
Патогенез:
Механізм розвитку МВ пов’язаний з дефектами синтезу білка, що виконує роль хлоридних каналів та бере участь у водно-електролітному обміні епітеліальних клітин дихальних шляхів, шлунково-кишкового тракту, підшлункової залози, печінки, репродуктивної системи. У зв’язку з нездатністю дефектного білка виконувати функцію хлоридного каналу, в середині клітини накопичуються іони хлору, змінюється електричний потенціал, сюди ж спрямовуються іони натрію, що обумовлює посилене всмоктування води з навколоклітинного простору. Внаслідок цього згущується секрет більшості залоз зовнішньої секреції, ускладнюється його евакуація, в органах виникають вторинні зміни, які найбільш виражені в бронхо-легеневій системі. В стінках бронхіального дерева розвивається хронічне запалення, формуються бронхіоло – і бронхоектази.
В умовах постійної обструкції в’язким харкотинням і зростаючої деструкції легеневої паренхіми бронхоектази стають розповсюдженими, наростає гіпоксія, розвивається легенева гіпертензія і, так зване, “легеневе серце”.
Бронхолегеневі зміни переважають в клінічній картині МВ і визначають прогноз у 95% пацієнтів. Проте МВ являє собою системне захворювання з ушкодженням шлунково-кишкового тракту і, як наслідок – серця та інших життєво важливих органів.
Клінічна картина:
Виразний поліморфізм МВ визначає різні варіанти його перебігу. У дітей перших місяців життя МВ можна запідозрити при наявності вроджених ателектазів, лобарної пневмонії, бронхіту з сухими та вологими хрипами (більше 2-3 тижнів), бронхіоліту, кашлю протягом більше, ніж 2-3 тижнів, навіть при відсутності хрипів в легенях.
В 8 – 20% випадків МВ маніфестує з меконіального ілеуса з можливим переходом в меконіальний перитоніт. В менш трагічних випадках випорожнення дуже об’ємні, з сильним неприємним запахом, “жирні”. В 1/3 хворих спостерігається випадіння прямої кишки, але при призначенні адекватної дози травних ферментів загроза анального пролапсу незначна.
Для дітей шкільного віку характерні кишкові коліки, здуття живота, повторна блювота; при обстеженні таких дітей в черевній порожнині пальпуються щільні “утвори”.
В деяких випадках розвивається симптоматика бронхіальної астми, при детальному обстеженні виявляється невелика ядуха, збільшення об’єму грудної клітки (в основному за рахунок передньо-заднього розміру). Незначне, але постійне зменшення екскурсії грудної клітки. Як наслідок перевантаження малого кола кровообігу розвивається “легеневе серце”, а хронічна гіпоксія, в свою чергу, призводить до деформації кінцевих фаланг пальців по типу “барабанних паличок” і нігтів по типу “годинникових скелець”.
Ураження підшлункової залози обумовлюється синдромом мальабсорбції з дистрофією та “жирними” випорожненнями у великій кількості. На пізній стадії розвивається цукровий діабет.
У 13% хворих змішаною та кишковою формами МВ розвивається цироз печінки (типовий для мутації W1282X). В половині випадків виявляють біліарний цироз з портальною гіпертензією. Загалом клінічні, лабораторні, інструментальні і морфологічні зміни печінки знаходять у 86% хворих.
У зв’язку з тим, що ушкоджуються всі органи, які мають слизопродукуючі залози, типовим є колітичний синдром, хронічний холецистит, сінусити.
Діагностичний критерій МВ у новонароджених – відсутність прибавки маси тіла до 14 днів від народження:
- Критеріями діагностики МВ в грудному віці є симптоми меконіальної кишкової непрохідності (відсутність випорожнень, блювота з жовчю, здуття живота, перітонеальні симптоми, погіршення загального стану), респіраторний синдром, гіпотрофія 2 – 3 ступенів (незважаючи на підвищений апетит), синдром мальабсорбції (особливо при переводі дитини на штучне та змішане вигодовування).
- Загальними симптомами, по яких можна запідозрити МВ, є дистрофія і відставання у фізичному розвитку, рецидивуючі хронічні захворювання органів дихання, поліпи носа, стійкий хронічний гайморит, хронічний бронхіт, рецидивуючий панкреатит, синдром мальабсорбціЇ з “жирними” випорожненнями, диспепсичні порушення неясної етіології, кровохаркання, цироз печінки, азооспермія, зниження фертильності у жінок, дихальна недостатність.
- Найбільш доступним для діагностики МВ є потовий тест (іонофорез з пілокарпіном). Підвищення хлоридів поту в концентрації більше 100 мекв/д обумовлює вірогідний діагноз; більше 40 мекв/д – для грудних дітей, більше 60 мекв/д – ймовірний. Для постановки остаточного діагнозу пілокарпінова проба має бути позитивною не менше трьох раз і, при цьому, завчасно необхідно виключити заняття спортом та будь-яке інше, що призводить до підвищення потовиділення.
Потову пробу необхідно проводити кожній хронічно кашляючій дитині.
- Для діагностики МВ має значення визначення хімотрипсину у випорожненнях. Для його визначення необхідно не менше, ніж за 3 дні до обстеження відмовитись від прийому травних ферментів. При МВ концентрація хімотрипсину знижена (ця проба нестандартизована, нормативні значення розробляються в конкретній лабораторії). В даному випадку диференційна діагностика проводиться з екзокринною панкреатичною недостатністю будь-якого генезу, синдромом Швахмана, станом після резекції шлунку по Більрот ІІ, білковим дефіцитом.
- При МВ визначають кількість жирних кислот у випорожненнях. Проміжне значення – 20-25 ммоль/день (в нормі менше 20 ммоль/день). Проба позитивна при зниженні екзокринної функції не менше, ніж на 75%. Диференційна діагностика проводиться з дефіцитом кон’югованих кислот в тонкому кишківнику (як умова емульгації жирів з їх наступною ферментацією ліпазою), печінковою недостатністю, непрохідністю жовчних шляхів, бактеріальною контамінацією тонкого кишківника з посиленим гідролізом жовчних кислот, з ілеітом, інтестінальними лімфомами, хворобою Уіпля, синдромом мальабсорбції при целіакії, харчовою алергією, прискореним пасажем їжі при діареях, карциноїдному синдромі, гіпертиреозі.
- Найбільш чутливою та специфічною є ДНК діагностика. Помилкові результати отримуються в 0,5-3% випадків. ДНК діагностика МВ виправдана для тих країн, де частота дельта – 508 вище 80%. Вважається, якщо жодна із 10 “локальних” мутацій не виявляється на тій чи іншій алелі хромосоми дитини, а шлюб батьків не кровноспоріднений, ймовірність МВ у даного пацієнта практично незначна. ДНК-обстеження батьків та інших членів родини без клінічних ознак хвороби недоцільне, за винятком випадків пренатальної діагностики.
- Пренатальна ДНК-діагностика можлива на основі дослідження навколоплідних вод, тканин плаценти в ІІ триместрі вагітності та є необхідною у зв’язку з високим ризиком повторення народження хворої дитини в сім’ї (25%).
- Проведення масового скрінінгу затруднене у зв’язку з тим, що в даний час ідентифікації доступно біля 75% мутантних хромосом, але можливе проведення каскадного скрінінгу, коли в центрі обстеження знаходиться родина хворого.
Лікування:
Лікування МВ потребує комплексного підходу і повинно включати в себе:
- застосування засобів, що покращують дренажну функцію бронхів;
- антибактеріальну терапію (постійні профілактичні курси);
- протизапальні засоби;
- замісну ферментотерапію.
Харчовий раціон повинен перевищувати вікові норми калоражу на 10-15%, на початкових етапах необхідне обмеження жирів, але при адекватній замісній ферментотерапії ця вимога не звучить так категорично. Обов’язкове введення полівітамінів, мікроелементів.
Передусім проводиться корекція лікувально-реабілітаційного режиму. Необхідно ефективно очищувати бронхіальне дерево від в’язкого харкотиння, боротися з інфекцією та забезпечити нормальний фізичний розвиток дитини.
Кінезотерапія включає постуральний дренаж, позиційний масаж, клопф-масаж, але ефективніше застосування форсованої експіраторної техніки, аутогенного дренажу.
При перших симптомах активації запалення призначають антибіотики курсом по 2-3 тижня.
Необхідним атрибутом терапії МВ є муколітики, які призначають як per os, так і в інгаляціях (ацетилцистеїн 50-200 мг 3 рази на добу).
Нормальний нутритивний статус – важлива мета в лікуванні дітей з МВ. Тому необхідна постійна ферментотерапія. Ефективні (по наростаючій) панкреатин, мезим-форте, ферменти з РН-чутливою оболонкой – креон, котазим, панцитрам.
Новими напрямками в лікуванні МВ є:
- застосування нестероїдних протизапальних препаратів (ібупрофен);
- альтернуючі курси кортикостероїдів (у дітей раннього віку з клінічно вираженою бронхіальною обструкцією);
- амілорід, блокатор натрію, АТФ, які відкривають альтернативні хлоридні канали, антитрипсин, сироватковий лейкоцитарний інгібітор протеаз.
Можна виділити ступені комплексного лікування МВ в залежності від рівня фінансування:
- Фундаментом успішної терапії є дієта, ферменти, вітаміни, фізіотерапія, чисте повітря, взаємодія в асоціації “лікар – сім’я – хворий”.
- Хороший контроль астми, звичайні антибіотики.
- Специфічні антибіотики внутрішньовенно та в інгаляціях.
- ДНК-аза і амілорід.
- Трансплантація легень і генна терапія.
Диспансерний нагляд за хворими на МВ включає:
- 1 раз в 3 місяці – антропометрія, дослідження функцій зовнішнього дихання, загальний аналіз крові та сечі, копрограма, аналіз харкотиння на флору та чутливість до антибіотиків;
- не рідше 1 разу в рік – рентгенографія органів грудної клітки, визначення кісткового віку, біохімічні та імунологічні дослідження, ехокардіографія, ультразвукове дослідження.
Формою активного спостереження є денний стаціонар.
Поради для батьків:
- Різні продукти потребують різної кількості ферментів для перетравлювання. Важливо додатково приймати ферменти, коли дитина їсть жирну їжу. Але крохмаль, білки та жиророзчинні вітаміни також потребують певної кількості ферментів. Найкращій спосіб дізнатися, чи достатню дозу ферментів приймає дитина – подивитися, чи не вздутий у неї живіт, чи не болить в неї бік, чи немає в неї об’ємних випорожнень з неприємним запахом. Проте тільки лікар визначить кількість капсул ферментів, необхідних для засвоєння їжі.
- Ферменти необхідно приймати одночасно з їжею. При цьому гранули не слід розжовувати.
- Ферменти особливо важливі, коли дитина захворіла. Наприклад, при загостренні бронхо – легеневого процесу, коли необхідно підтримувати енергетичний баланс. Іноді, коли дитина захворіла, і їй не хочеться їсти, можуть бути дуже корисні висококалорійні напої та молоко.
- Велике значення має фізіотерапія. Вона починається з дихальних вправ, які мають бути двох типів:
- Контроль дихання – при вдосі животик дитини повинен значно збільшитися. Далі видихнути так, щоб він набув звичайних розмірів. Робити вдохи та видохи треба правильно.
- Глибоке дихання – зробити глибокий вдих, щоб нижня частина грудної клітки збільшилась та розширилась. Після цього видихнути так, щоб грудна клітка була звичайних розмірів.
- Цикли активного дихання проводяться в порціях постурального дренажу (чи нахильного положення) – верхня середина ліва та права, нижня ліва та права.
Таким чином, об’єднані зусилля педіатрів, терапевтів, пульмонологів, гастроентерологів, генетиків, дієтологів, психологів, за умови ранньої діагностики та адекватної терапії вже на сучасному етапі суттєво змінюють прогноз МВ.
Номер з каталогу МІМ:
- 602421 Cystic Fibrosis Transmembrane Conductance Regulator; CFTR.
- 219700 Cystic Fibrosis; CF.
- 603855 Cystic Fibrosis Modifier 1; CFM1.
- 219721 Cystic Fibrosis with Helicobacter Pylori Gastritis, Megaloblastic Anemia, and Mental Retardation.
Літературні джерела:
- Andersen DH. Cystic fibrosis of the pancreas and its relation to celiac disease. A clinical and pathologic study. Am J Dis Child 1938;56:344-99.
- Cystic Fibrosis Foundation. 1995. Patient Registry 1994 Annual Data Report. Bethesda, Maryland.
- Cystic Fibrosis Trust. Management of cystic fibrosis in adults. UK. 1995.
- Davis PB, et al. Cystic Fibrosis. Am J Respir Crit Care Med 1996;154:1229-56.
- Govan JWR, et al. Evidence of transmission of Pseudomonas cepacia by social contact in cystic fibrosis. Lancet 1993;342:15.
- Hodson ME. Aerosolized dornase alfa (rhDNase) for therapy of cystic fibrosis. Am J 1995;151:70-4.
- Konstan MW, et al. Effect of high-dose ibuprofen in patients with cystic fibrosis. 1995.
- Kotloff, Zuckerman. Lung transplantation for cystic fibrosis: special considerations. Chest March 1996;109(3):787-98.
- Niolaizik WH, MH. Schoni. Pilot study to assess the effect of inhaled corticosteroids on lung function in patients with cystic fibrosis. J Pediatr 1996;128:271-4.
- Pryor JA and Webber BA. Physiotherapy for cystic fibrosis-which technique? Physiotherapy 1992;78:105-8.
- Southern KW. Gene therapy for cystic fibrosis: current issues. Brit J Hosp Med 1996;55(8):495-9.
- Taussig LM. The reproductive system. In: Cystic Fibrosis /Brugman, S. M. (Еd. L. M. Taussig), Thieme-Stratton, New York 1984;324-7.
- Van Haren EHJ, et al. Bronchodilator response in adult patients with cystic fibrosis: effects on large and small airways. Eur J Respir Dis 1991;4:301-7.
- Warner JO, et al. Cystic fibrosis in children, in Respiratory Medicine (second edition), London 1995:1330-40.
- Webb AK. The treatment of pulmonary infection in cystic fibrosis. Scand J Infect Dis Suppl 1995;96:24-7.
- Капранов Н. И. Рачинский С. В. Муковисцидоз.- М, 1995.
- Петрова Н. В. Материалы научно-практической конференции РДКБ. – М, 1995.- C. 96.
- Потапова О. Ю. Автореферат – Молекулярно-генетический анализ кистозного фиброза в России – Спб, 1994.- С. 24.
- Чучалин А. Г., Самильчук Е. И. Муковисцидоз – состояние проблемы //Тер. архив. – 1993.- № 65 (3).- C.3-8.
Переглянуто редакційною колегією I.B.I.S.: 15/02/2002
Дивіться також:
- Діагностика, лікування та профілактика муковісцидозу (методичні рекомендації)
Об’єм та довжина яєчок
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”
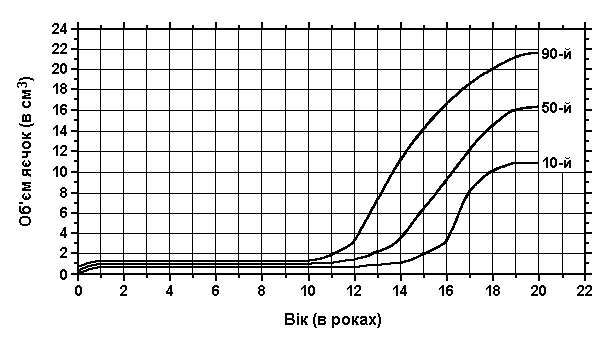
Дані отримані в результаті обстежень 1500 хлопчиків та чоловіків (перехресне популяційне дослідження в різних лікарнях та різних містах). Schonfield WA: Am J Dis Child 65:535, 1943.
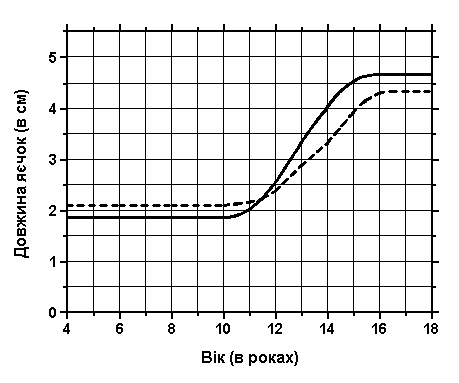
Повздовжній ріст яєчок адаптований з нормальних стандартів об’єму яєчок. Суцільною лінією подані дані з A.Prader, Zurich; пунктирна лінія – це дані з Laron A та Zilka E: J Clin Endocrinol Metab 29:1409, 1969 – адаптовано з Smith DW: Recognizable Patterns of Human Malformation, ed. 3, W.B.Saunders, 1982.
Розмір пенісу
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”
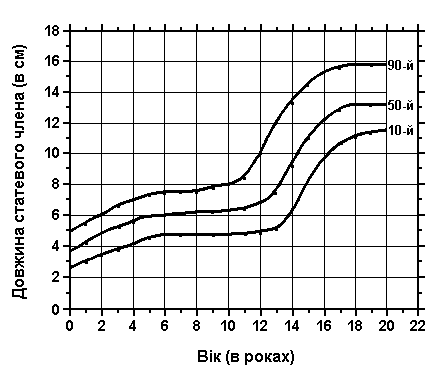
Ріст пенісу – повна розтягнута довжина від лобкової кістки до голівки – вимірювався від дитинства до дорослого віку. Дані отримані в результаті обстежень 1500 хлопчиків та чоловіків (перехресне популяційне дослідження в різних лікарнях та різних містах). Schonfield WA: Am J Dis Child 65:535, 1943.
Мікроцефалія
(Microcephaly)
Л.С. Євтушок
Зав. медико-генетичною консультацією
Рівненського клінічного лікувально-діагностичного центру
Визначення:
Визначення мікроцефалії не є однозначним. Якщо за критерій взяти потилично – лобний обвід голови, що на 2 або більше стандартних відхилення (СВ) менший від норми для цього віку та статі, то приблизно 2,3% усього населення будуть мікроцефалами; при цьому лише невелика його частка буде мати неврологічні розлади. Якщо взяти за критерій мікроцефалії величину потилично-лобного обводу голови, який на 3 або більше СВ нижчий за норму для даного віку та статі, тоді лише у 0,1% пацієнтів буде визначено мікроцефалію, проте в цій групі буде сильніше виражена неврологічна симптоматика та розумова відсталість. З точки зору педіатрів критерієм мікроцефалії краще вважати 3 СВ.
Однак слід пам’ятати, що крім розмірів голови, зазначених в таблицях Нелхауза існують дані про різні національні норми.
Етіологія:
Мікроцефалія може бути спричинена різними хромосомними аномаліями, моногенними порушеннями, пре, – інтра – та постнатальними причинами. Саме тому можливі різноманітні клінічні прояви. Деякі автори поділяють мікроцефалію на дві великі групи: первинна та вторинна мікроцефалія. При первинній мікроцефалії, зазвичай, немає інших мальформацій: вона відповідає менделевським принципам успадкування або асоціюється з певним генетичним синдромом. Вторинна мікроцефалія може бути результатом великої кількості чинників, які могли вплинути на плід або дитину в період швидкого розвитку мозку, особливо протягом перших 2 років життя.
Приклади первинної мікроцефалії:
Родинна мікроцефалія. Аутосомно-рецесивний тип успадкування. Частота 1:40000 новонароджених. Типовий прояв – скошене чоло, відстовбурчені вуха, середній ступінь розумової відсталості. Приблизно 1/3 хворих має приступи судом, які важко контролювати. Тривалість життя може бути нормальною в неускладнених випадках. Вважається, що 1 із 100 індивідів в популяції є носієм аутосомно -рецесивного гену мікроцефалії.
Мікроцефалія із аутосомно-домінантним типом успадкування. Зазвичай на обличчі не має характерних проявів, може бути незначно скошене чоло, відстовбурчені вуха. Розумова відсталість середнього чи важкого ступеня. Якщо виникають судоми, то їх легко контролювати.
Синдроми: Дауна, Трисомії 18, Трисомії 13, делеції 5p (кошачого крику), Брахмана-деЛанге, Рубінштейна-Тейбі, Сміта-Лемлі-Опітца.
Приклади вторинної мікроцефалії:
Вплив опромінювання на плід. Мікроцефалія та розумова відсталість найбільш виражені, якщо опромінювання відбувалося протягом перших 15 тижнів гестації.
Вроджені інфекції: цитомегаловірус, краснуха, токсоплазмоз.
Наркотики, алкоголь, ліки: фетальний алкогольний синдром, фетальний гідантоіновий синдром, інші наркотики, такі, як ізотретинова кислота, кокаїн.Порушення, пов’язані з тетрагідробіоптеріном.
Інші пренатальні впливи: материнська ФКУ (гіперфенілаланінемія); внутрішньоутробна асфіксія.
Допологові / післяпологові впливи: гіпоксично-ішемічна енцефалопатія, менінгіт або енцефаліт, деякі вроджені порушення метаболічних процесів, дефіцит піруватдегідрогенази, некетонна гіперглікемія).
Діагностика:
- Регулярні вимірювання, що зображаються графічно. Якщо виявлене зменшення обводу голови для даного віку і статі, доцільно провести кілька контрольних вимірів в динаміці через певні проміжки часу. Вимірювання розмірів голови відразу після народження та через кілька місяців є важливим для диференціювання гіпоксично – ішемічного ураження мозку (нормальні розміри голови при народжені) від первинної пренатальної мікроцефалії (маленькі розміри голови при народженні). Перинатальна гіпоксія не є причиною вродженої мікроцефалії. Мікроцефалія, діагностована пізніше, може бути проявом деяких метаболічних розладів, інфекцій центральної нервової системи або крововиливів в мозок та таких синдромів, як синдром Ретта і синдром Ангельмана.
- Повторна оцінка розмірів обводу голови у випадках набряку скальпа та кефалогематоми. Ще однією діагностичною помилкою періоду новонародженості є “набута мікроцефалія” – деформація черепа, однією з причин якої є дворога матка.
- Краніосиностоз (передчасне змикання черепних швів) має бути виключеним. Зазвичай в таких випадках спостерігається деформація черепа, яка є прямим наслідком передчасного змикання швів. Пальпація цих швів дозволяє виявити виступаючі краї кісток. Змикання швів можна діагностувати при рентгенографії черепа або комп’ютерній томографії у неясних випадках.
- По можливості, провести вимірювання розмірів черепа кожного з батьків та братів / сестер. Це допоможе встановити випадки родинної мікроцефалії.
- Треба провести генеалогічний аналіз на предмет повторення випадків мікроцефалії або вад, пов’язаних із центральною нервовою системою.
- Переглянути історію пологів на предмет виявлення таких захворювань матері, як діабет або внутрішньоутробні інфекції, вживання алкоголю, прийом медикаментів, прийом заборонених медикаментів, опромінювання, фетоплацентарна недостатність або будь-які інші ускладнення під час вагітності.
Клінічне обстеження:
- Пошук будь-яких інших вад розвитку, дизморфічних ознак, порушень розвитку;
- Пошук аномалій очей: мікрофтальмії, хоріоретиніту, катаракти та інших;
- Визначення наявності розумової відсталості і її ступеню;
- Виявлення неврологічних розладів (яскраво виражених чи прихованих);
- Синдромологічний аналіз.
Генетичний висновок:
Генетична консультація – необхідний аспект при комплексному обстеженні хворого на мікроцефалію, оскільки ризик повторення її в сім’ї залежить від генетичного діагнозу. Діагностична програма при мікроцефалії включає клінічне обстеження, включаючи огляд офтальмолога, невропатолога, хромосомний аналіз, дослідження порушень метаболізму, відповідно до кожного випадку. Результатом генетичного обстеження стає визначення типу мікроцефалії: Випадки “чистої” (неускладненої) мікроцефалії (без спастичності, судом чи аномалій ока):
- Якщо ступінь розумової відсталості є середнім, то у пацієнта аутосомний ген з мінливою експресивністю. Не забувати провести вимірювання обводів голови батьків та братів / сестер. У випадках аутосомно-домінантої мікроцефалії – коли один з батьків є хворим, ризик передачі гена будь-кому з нащадків становить 50%.
- Якщо ступінь розумової відсталості є більш важким з малим, але нормальної структури мозком за даними неврологічних тестів, то найбільш вірогідним буде діагноз аутосомно – рецесивної мікроцефалії. Ризик повторного народження дитини з мікроцефалією – 25%.
- У випадках “чистої” мікроцефалії без розумової відсталості вірогідним є діагноз мікроцефалії із схильністю до злоякісних пухлин лімфоретикулярної системи, синдрому Ніхмегена. Хромосомний аналіз дозволяє виявити підвищення рівня спонтанних розривів і перебудов.
Мікроцефалія з приступами судом та спастикою:
- Оцінка неврологічного статусу може дати ключ до діагнозу, встановивши аномальну мієлінізацію, кальцинати, голопрозенцефалію, інші структурні аномалії чи гіпоксично-ішемічні ураження;
- З іншого боку, пацієнт вважається носієм аутосомно-рецесивного гена, доки не буде доведене інше (зазвичай у випадках, коли окружність голови менша за 3 СВ відповідно до віку та статі).
- Випадки Х-зчепленої мікроцефалії дуже рідкісні.
Мікроцефалія з аномаліями ока:
- Існує декілька синдромів мікроцефалії та аномалій ока. Успадкування таких випадків відбувається відповідно до законів Менделя.
- В деяких випадках визначалась мікроцефалія з хоріоретинопатією (псевдотоксоплазмоз). Вони були задокументовані як випадки з аутосомно-домінантним та аутосомно-рецесивним успадкуванням.
- У випадках мікрофтальму або анофтальму, що асоціюються з мікроцефалією (дисплазія Ленца), успадкування відбувається Х-зчеплено.
Мікроцефалія та дизмофічні ознаки:
- У пацієнта має бути розпізнаний синдром. В цьому може допомогти інформаційна база даних по дизморфології.
- Якщо в пацієнта виявляють дизморфії та мікроцефалія в межах від 2 до 3 СВ, але нема підстав вважати це спадковим фенотипом (ідіопатична мікроцефалія або мікроцефалія невідомої етіології), то емпіричний рекурентний ризик приблизно 5%, що близько до рекурентного ризику, який визначається для неуточнених дизморфічних синдромів.
Прогноз:
- Буде залежати від багатьох факторів, включаючи ступінь розумової відсталості, наявність інших порушень розвитку та судом.
- Пацієнти із аутосомно-домінантною мікроцефалією зазвичай мають нормальну тривалість життя.
- Пацієнти із аутосомно-рецесивною мікроцефалією можуть мати нормальну тривалість життя, якщо в них нема корчів або їх легко контролювати.
Пренатальний діагноз
Незважаючи на те, що при УЗД плода можна виявити випадки церебрального дизгенезу або зменшений міжтім’янний діаметр для даного терміну гестації, приблизно 40-50% випадків мікроцефалії виявляються у третьому триместрі або вже після народження.
Діагностичні тести:
- Детальне офтальмологічне обстеження може дати привід для визначення певного окуло-церебрального синдрому.
- Неврологічне обстеження включає комп’ютерну томографію, магнітно-резонансне сканування мозку черепа, необхідні для визначення діагнозу для пацієнтів із структурним ураженням мозку. Наприклад, у випадках, коли розміри мозку зменшені, але він правильно сформований, або для виключення голопрозенцефалії, кальцинатів чи аномальної мієлінізації. Необхідно пам’ятати, що внутрішньочерепні кальцинати спричинені не лише внутрішньоутробними інфекціями. Гіпоксично-ішемічне ушкодження та аутосомно-рецесивний синдром, що асоціюється із хоріоретінітом, можуть мати подібні прояви. Рентгенографія черепа у прямій проекції дозволить виявити більшість випадків краніосиностозу. Комп’ютерне сканування необхідне у сумнівних випадках передчасного змикання швів та ідентифікації внутрішньочерепних кальцинатів. Магнітно-ядерний резонанс необхідний у випадках, коли є очевидні прояви дизморфогенезу центральної нервової системи.
- Лабораторні тести призначаються відповідно до висновків після вивчення історії хвороби та фізичного огляду. Зазвичай вони включають обстеження на TORCH-інфекції, серологічні докази внутрішньоутробної інфекції, особливо токсоплазмозу, цитомегаловірусу, краснухи, герпесу. Серологічні або мікробіологічні проби на внутрішньоутробні інфекції необхідно проводити протягом перших місяців життя.
- Хромосомний аналіз необхідний при мікроцефалії з множинними дизморфічним ознаками, так і при мікроцефалії без інших дизморфічних ознак, оскільки вона може бути спричинена хромосомним дисбалансом та кільцевими хромосомами.
- Дослідження метаболізму. Якщо причина мікроцефалії невідома, необхідно визначити рівень фенілаланіну в сироватці крові матері, оскільки материнська ФКУ може викликати мікроцефалію у дитини, навіть якщо у матері немає ніяких симптомів, а дитина не має ФКУ. Якщо на основі клінічної картини хворого запідозрені метаболічні порушення, то можна порадити аналізи для визначення дефіциту піруватдегідрогенази, некетонної гіпоглікемії, чи атипової метілмалонової ацидурії. Такі тести включають визначення спектру амінокислот в плазмі крові, органічних кислот в сечі, пірувату в крові та рівню лактату, рівня цукру в крові.
Лікування:
Якщо причина (етіологічний діагноз) мікроцефалії встановлена, лікар повинен дати відповідні поради щодо генетики даного випадку і родинного прогнозу. Незважаючи на те, що специфічне лікування мікроцефалії неможливе, існують програми, які допомагають дітям із розумовою відсталістю максимально реалізувати їхні можливості в розвитку. Крім того, існують спеціальні медичні поради, що можуть застосовуватись для певних синдромів, що асоціюються із мікроцефалією (синдром Дауна, Корнелії де Ланге). Ці поради мають допомогти лікарям та іншим медпрацівникам підтримувати дітей із специфічними хворобами та покращувати їх життя шляхом своєчасного встановлення діагнозу та лікування, увагою до їх психологічних потреб.
Література:
- Brandon, M. G. W.; Kirman, B. H.; Williams, C. E.: Microcephaly in one of monozygous twins. Arch. Dis. Child. 34: 56-59, 1959.
- Cowie, V.: The genetics and sub-classification of microcephaly. J. Ment. Defic. Res. 4: 42-47, 1960.
- Davies, H.; Kirman, B. H.: Microcephaly. Arch. Dis. Child. 37: 623-627, 1962.
- Ferguson-Smith, M. A.: Personal Communication. Glasgow, Scotland 7/9/1981.
- Hanhart, E.: Ueber einfache Rezessivitaet bei Mikrocephalia vera, spuria et combinata und das herdweise Vorkommen der Mikrocephalia vera in Schweizer Isolaten. Acta Genet. Med. Gemellol. 7: 445-524, 1958.
- Jackson, A. P.; McHale, D. P.; Campbell, D. A.; Jafri, H.; Rashid, Y.; Mannan, J.; Karbani, G.; Corry, P.; Levene, M. I.; Mueller, R. F.; Markham, A. F.; Lench, N. J.; Woods, C. G.: Primary autosomal recessive microcephaly (MCPH1) maps to chromosome 8p22-pter. Am. J. Hum. Genet. 63: 541-546, 1998.
- Kloepfer, H. W.; Platou, R. V.; Hansche, W. J.: Manifestations of a recessive gene for microcephaly in a population isolate. J. Genet. Hum. 13: 52-59, 1964.
- Koch, G.: Genetics of microcephaly in man. Acta Genet. Med. Gemellol. 8: 75-86, 1959.
- Komai, T.; Kishimoto, K.; Ozaki, Y.: Genetic study of microcephaly based on Japanese material. Am. J. Hum. Genet. 7: 51-65, 1955.
- Mikati, M. A.; Najjar, S. S.; Sahli, I. F.; Melhem, R. E.; Mansour, S.; Der Kaloustian, V. M.: Microcephaly, hypergonadotropic hypogonadism, short stature, and minor anomalies: a new syndrome. Am. J. Med. Genet. 22: 599-608, 1985.
- Perez-Castillo, A.; Martin-Lucas, M. A.; Abrisqueta, J. A.: Is a gene for microcephaly located on chromosome 1? Hum. Genet. 67: 230-232, 1984.
- Plummer, G.: Anomalies occurring in children exposed in utero to the atomic bomb in Hiroshima. Pediatrics 10: 687-693, 1952.
- Qazi, Q. H.; Reed, T. E.: A possible major contribution to mental retardation in the general population by the gene for microcephaly. Clin. Genet. 7: 85-90, 1975.
- Qazi, Q. H.; Reed, T. E.: A problem in diagnosis of primary versus secondary microcephaly. Clin. Genet. 4: 46-52, 1973.
- Rizzo, R.; Pavone, L.: Autosomal-recessive microcephaly in two siblings, one with normal IQ and both with protruding mandible, small ears, and curved nose. Am. J. Med. Genet. 59: 421-425, 1995.
- Tolmie, J. L.; McNay, M.; Stephenson, J. B. P.; Doyle, D.; Connor, J. M.: Microcephaly: genetic counselling and antenatal diagnosis after the birth of an affected child. Am. J. Med. Genet. 27: 583-594, 1987.
- Van den Bosch, J.: Microcephaly in the Netherlands: a clinical and genetical study. Ann. Hum. Genet. 23: 91-116, 1959.
Переглянуто редакційною колегією I.B.I.S.: 05/02/2002
Статевий розвиток хлопчиків (продовження)
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”
Статевий розвиток – ріст геніталій в чоловіків
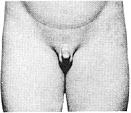
Стадія 1. Пеніс, яєчка та калитка дитячого розміру.
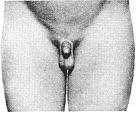
Стадія 2. Спостерігається збільшення калитки та яєчок. Пеніс, зазвичай, не збільшується. Шкіра на калитці червоніє.
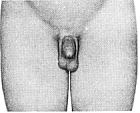
Стадія 3. Спостерігається подальший рост яєчок та калитки і збільшення пенісу, переважно його довжини.
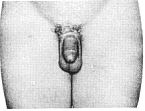
Стадія 4. Подальший ріст яєчок та калитки, збільшення пенісу, особливо його ширини.
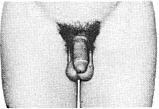
Стадія 5. Статеві органи мають дорослі розміри та форму..
Статевий розвиток – ріст лобкового волосся
Стадія 1. Відсутність лобкового волосся.
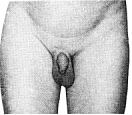
Стадія 2. Спостерігається невеликий ріст довгого, трохи пігментованого, прямого або злегка курчавого волосся, переважно в основі пенісу.
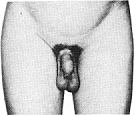
Стадія 3. Волосся стає помітно темнішим, жорсткішим та більш курчавим. Волосся повільно поширюється по лобку.
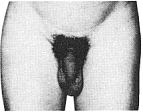
Стадія 4. Тепер волосся є дорослим за структурою, але вкриває меншу площу і не поширюється на внутрішню сторону стегон.
Стадія 5. Волосся відповідає дорослому віку за своєю структурою та кількістю і поширюється на стегна.
Ілюстрації подані за згодою J.M. Tanner. Tanner JM: Growth at Adolescence, ed. 2, Blackwell Scientific Publications, 1982.
Мікротія, атрезія
(Microtia, Atresia)
Т.В. Віговська
Зав. неонатологічним центром
Волинського обласного дитячого територіального медичного об’єднання
Включені:
Aнотія, дизгенезія вуха, мікротія – атрезія, мікротія – атрезія меатуса – кондуктивна глухота, кістково-провідникове зниження слуху, гіпогенезія вушної раковини, асоційована з атрезією зовнішнього вуха, талідомід – обумовлена мальформація зовнішнього вуха.
Виключені:
Aллопеція – аносмія – глухота – гіпогонадизм типу Джонсонівського; атрезія зовнішнього слухового ходу, ізольована бранхіо-ото-ренальна дисплазія; глухота-дефект-вуха-лицьовий параліч; вухо, крипнотія, мальформація зовнішнього вуха, ізольована; посттравматична чи післяхірургічна деформація вушної раковини; стеноз зовнішнього слухового проходу; аурикулярна мальформація, інші.
Основні діагностичні критерії:
Деформована або відсутня вушна раковина з атрезією слухового ходу, з основними формами кондуктивного зниження слуху.
Клінічна картина:
Праве вухо пошкоджується частіше лівого; 1/6 пацієнтів має двостороннє пошкодження. В деяких випадках мікротія з атрезією або без неї може спостерігатись на одній стороні і атрезія ізольована на протилежній. Вушна раковина може бути тільки дещо менших розмірів і мати конфігурацію нормального вуха. В більшості випадків вушна раковина є скрученою і значно деформованою, з декількома хрящовими залишками. Періодично, зовнішнє вухо відсутнє повністю (анотія). Зовнішній слуховий канал може бути відсутнім, звужений на всьому протязі, або бути лійкоподібно звуженим аж до повної атрезії за декілька міліметрів до конхії. Іноді маленький отвір можна знайти спереду, вище або позаду по відношенню до нормального розміщення слухового ходу. Часто цей отвір закінчується сліпо, але зрідка сполучається з барабанною перетинкою. Трапляється, що деякі з цих деформованих каналів містять сірчані залози, і сірку можна виявити у просвіті. Можуть бути деформованими різні частини вуха. В одних випадках відсутній гелікс, в інших – деформовані мочки вуха. Можуть спостерігатися різні ступені деформації трагуса в асоціації з преарикулярними додатками. Дуже рідко при анотії взагалі відсутня відстань між mastoidens і відростком нижньої щелепи, що необхідно для фукнціонування темпоро-мандибулярного суглоба. За таких умов аномалія середнього вуха може асоціюватись з гіпоплазією чи аплазією середнього вуха, що робить реконструкцію слухового механізму важкою чи неможливою. Мастоїдальний відросток може бути недорозвиненим. Іноді на місці барабанної порожнини хірурги знаходять м’яку тканину, але частіше присутня потовщена кісткова пластинка. У винятково рідких випадках барабанна перетинка лежить медіально від місця атрезії і це може асоціюватись з холестеатомою і значною деструкцією кістки. Майже завжди присутня кондуктивна приглухуватість з кістково-провідною до 40-60 децибел. У деяких випадках спостерігається і нейросенсорна приглухуватість, обумовлена супутньою аномалією внутрішнього вуха. Вестибулярні порушення звичайно не спостерігаються. Рентгенодослідження з використанням високочутливої комп’ютерної томографії середнього і внутрішнього вуха є необхідним для планування хірургічного втручання і прогнозування. Якщо рентгенодослідження виявляє виражену деформацію чи аплазію середнього вуха, хірургічне втручання може не мати сенсу. Рентгенодослідження може також показати атипове положення n. facialis в висковій кістці, часто асоційовану аномалію. Щілина середнього вуха і pr. mactoideus можуть бути нормальних розмірів чи гіпоплазовані. Молоточок і коваделко часто деформовані або прифіксовані до кісткової основи. Стрім’ячко може бути деформоване або прифіксоване до основи, з порушенням інкостапедіального з’єднання і відсутністю округлого вікна.
Ускладнення:
Врата слухопровідності відповідно до атрезії чи асоціації з аномалією середнього вуха. Частота приблизно 100%. Зміщення, аномалія чи гіпоплазія лицьового нерва з частковим або повним паралічем. При оперативному втручанні можна знайти середній отит чи мастоїдит.
Поєднані симптоми:
Гіпоплазія латеральної частини нижньої щелепи та обличчя відповідно аномалій комплексу першої зябрової дуги; звуження кісткової частини євстахієвої труби, відсутність torus tubarius (дуже рідко), ненормальне розширення євстахієвої труби. Відсутність або гіпоплазія g. parotis, та відповідних мигдаликів. Преаурикулярні ямки та заглибини, бранхіальні цисти, гіпоплазія та зміщення m. tensor tympani, m. tendor tympani, розщелина піднебіння, тератома мигдалика, різноманітні черепно-лицьові, скелетні вади та аномалії внутрішніх органів.
Етіологія:
Несинодромна (ізольована) мікротія є результатом аутосомно-домінантного наслідування в деяких сім’ях. Спорадично спостерігається у сібсів від нормальних батьків. Більшість випадків не ізольовані. У деяких батьків були знайдені 18q- і 18 трисомії. Талідомід викликає мікротію і анотію з паралічем черепних нервів і аплазією внутрішнього вуха. Може обумовлюватись вірусом краснухи та іншими внутрішньоутробними інфекціями.
Патогенез:
Порушення розвитку першої зябрової щілини, горбочка вушної раковини, які звичайно формуються із першої і другої зябрової дуги. Первинний канал, який формується із мезодермальної серцевини може реканалізуватись і сформувати розщелину піднебіння. Оскільки перша зяброва дуга розвивається неправильно, всі інші структури, що утворюються з неї (молоточок, коваделко, m. tensor tympani, mandibula) також можуть бути деформовані.
Друга зяброва дуга також може бути задіяна, що виразиться аномалією стрім’ячка і лицьового нерва.
Співвідношення статей: Ч > 1 : Ж 1.
Деякі дослідники вважають, що явне переважання випадків у чоловіків пояснюється і тим, що жінки ховають деформацію під волоссям.
Частота випадків:
1 : 10000, до 1 : 20000, серед індійців навахо 1 : 1200 живонароджених.
Ризик повторення для сибсів та дітей пацієнта:
Ізольована анотія наслідується по аутосомно-домінантному типу, повторний ризик 50%.
Вік маніфестації:
При народженні. Однак коли аномалія середнього вуха з вираженою кондуктивною глухотою має місце на фоні не зміненого слухового каналу, втрата слуху може бути довгий час не діагностована.
Генне картування і групи зчеплення генів: невідомі.
Профілактика: невідома.
Показане генетичне консультування.
Лікування:
Реконструкція зовнішнього, середнього вуха та слухового каналу. Якщо дефект двосторонній, до проведення реконструктивної операції слід використовувати слуховий апарат. Коли хірургічна корекція слуху не має сенсу, слід рекомендувати слухїові апарати, навчання на перших партах в школі та мовну терапію. При двосторонніх ураженнях хірургічне втручання на одній стороні слід проводити біля 5-річного віку, а втручання на протилежній стороні відстрочити до юнацьких років.
Прогноз:
Позитивний для життя за відсутності серйозних супутніх дефектів. Відновлення задовільного слуху спостерігається в 50-67%. У деяких пацієнтів після хірургічного втручання можуть розвинутись ускладнення у вигляді ураження лицьового нерва, стенозу слухового нерва. У деяких пацієнтів пошкодження внутрішнього вуха закінчується повною глухотою.
Діагностика носійства: невідома.
Зверніть увагу!
Дитина з мікротією-атрезією потребує ретельної оцінки анатомії зовнішнього каналу середнього та внутрішнього вуха. Слух має бути ретельно перевіреним, навіть коли немає атрезії. Для виключення аномалії середнього і внутрішнього вуха слід провести комп’ютерну томографію протилежного вуха, оскільки ці порушення можуть спостерігатись і там. Пацієнт також повинен бути ретельно оглянутий для виключення значних супутніх дефектів.
Літературні джерела:
- Bergstrom LV. Ear, microtia-atresia. In: Buyse ML, ed. Birth Defects Encyclopedia. Dover: Center for Birth Defects Information Services, Inc., 1990:591-592.
- Harris J, Kallen B, Robert E. The epidemiology of anotia and microtia. Journal of Medical Genetics 1996;33(10):809-813.
- Lammer EJ, et al. Retinoic acid embryopathy. New England Journal of Medicine 1985;313:837-841.
- Mastroiacovo P, et al. Epidemiology and genetics of microtia–anotia: a registry based Study on over one million births. Journal of Medical Genetics 1995;32:453-457.
- Stephan MJ. Autosomal recessive form of mandibular dysostosis. American Journal of Medical Genetics 1990:35:493-495.
- Warkany J. Aural defects. In: Congenital Malformations: Notes and Comments. Chicago: Yearbook Medical Publishers, Inc., 1971:401-416.
Переглянуто редакційною колегією I.B.I.S.: 05/02/2002
Синдром Стіклера
(Stickler Syndrome)
Жанна Мартинюк
Лікар-неонатолог
Волинського обласного дитячого територіального медичного об’єднання
Синоніми:
- Arthroophthalmopathy, hereditary progressive.
- Stickler syndrome, type I.
- Stickler syndrome, vitreous type I;
- Stickler syndrome, membranous vitreous type.
- Stickler syndrome, type II.
- Stickler syndrome, vitreous type II;
- Stickler syndrome, beaded vitreous type.
- Stickler syndrome, type III.
- Stickler syndrome, nonocular type.
Мінімальні діагностичні oзнаки:
- патологія очей;
- патологія скелету та суглобів;
- патологія внутрішнього вуха;
- аномалії лицевого черепа.
Етіологія:
Спадкове прогресуюче захворювання, що передається від батьків до дитини, викликане мутацією декількох генів, що відповідають за синтез колагену. Синдром названий на честь лікаря Ганнара Стіклера, який в 1960 році обстежував хлопчика, що мав костисті нарости суглобів, був низьким, його мати була сліпа, інші члени сім’ї мали подібні oзнаки.
Співвідношення статі: Ч1 : Ж1.
Частота виникнення: 1:10000.
Загальні клінічні прояви:
Період новонародженості:
- розщелини піднебіння;
- розщеплення язичка;
- наявність міопії, при нормі для новонароджених гіперметропії;
- екзофтальм;
- П’єра Робена синдром;
- лійкоподібна грудна клітка;
- арахнодактилія;
- збільшення, гіперрухливість суглобів;
- короткий ніс;
- розщелини хребта;
- різомелічне вкорочення кінцівок.
Дитинство:
- легка затримка росту;
- остеоартрит з підліткового віку, сколіоз, кіфоз, біль в суглобах;
- гіпоплазія тазових кісток, зниження осифікації шийки стегна;
- міопія, катаракта, глаукома, раптове відшарування сітківки, що веде до сліпоти;
- зниження слуху, отит;
- пролапс мітрального клапану.
Дорослий вік:
- артропатія (після 40 років), кульшових, колінних, гомілкових суглобів;
- відшарування сітківки, сліпота;
- зниження слуху, глухота;
- зменшення вираженності аномалій обличчя з віком.
Stickler syndrome, type I: 75% випадків.
Номер з каталогу МІМ: 108300 Stickler syndrome, type I; STL1.
Генне картування: мутація гена COL 2 А 1, локалізованого на 12 q 13. 11-q13.2.
Тип наслідування: аутосомно-домінантний з варіабельною експресивністю.
Симптоми:
- марфаноїдний фенотип;
- гіпоплазія середньої частини обличчя;
- нейросенсорна глухота;
- міопія;
- відшарування сітківки;
- сліпота, аномалії пігментації сітківки;
- катаракта;
- глаукома;
- вивернуті ніздрі;
- вдавлене перенісся;
- розщелини піднебіння;
- П’єра Робена синдром;
- пролапс мітрального клапана;
- лійкоподібна грудна клітка;
- легка спонділоепіфізарна дисплазія;
- платиспондилія;
- сколіоз, кіфоз;
- нерівномірне сплющення епіфізів стегнових кісток;
- арахнодактилія;
- артропатія.
Stickler syndrome, type IІ.
Номер з каталогу МІМ: 604841 Stickler syndrome, type II; STL2.
Генне картування: мутація COL 2 А 1, локалізованого на 1 р 21. 11-q13.2.
Тип наслідування: аутосомно-домінантний.
Симптоми:
- гіпоплазія середньої частини обличчя;
- мікрогнатія;
- нейросенсорна глухота;
- висока не прогресуюча міопія (старше 6 років);
- катаракта;
- глаукома;
- відшарування сітківки;
- вдавлене перенісся;
- вивернуті ніздрі;
- розщелини піднебіння;
- П’єра Робена синдром;
- розщеплення язичка;
- легка спонділоепіфізарна дисплазія;
- тонкі кінцівки, арахнодактилія;
- підвищена рухливість суглобів;
- артропатії після 30-40 років.
Stickler syndrome, type III.
Номер з каталогу МІМ: 184840 Stickler syndrome, type III; STL3.
Генне картування: мутація COL 2 А 2, локалізованого на 6 р 21.3.
Тип наслідування: аутосомно-домінантний.
Симптоми:
- гіпоплазія середньої частини обличчя;
- нейросенсорна глухота;
- відсутні очні симптоми;
- вивернуті ніздрі;
- розщелини піднебіння;
- П’єра Робена синдром;
- епіфізарна дисплазія;
- легка платиспондилія;
- біль в суглобах;
- ранній остеоартрит;
- великі епіфізи.
Обстеження:
Пренатально:
- біопсія ворсин хоріона в 10-12 тижнів вагітності;
- амніоцентез в 16-18 тижнів вагітності;
- УЗО 19-20 тижнів вагітності для виявлення розщелини піднебіння;
- молекулярно-генетичне консультування.
Період новонародженості:
- консультація щелепно-лицевого хірурга, окуліста, ортопеда, генетика.
В старшому віці:
- аудіограма;
- огляд окуліста, кардіолога;
- огляд ортопеда, Rо-графія кісток, біопсія синовіальної оболонки.
Диференціальний діагноз:
Stickler syndrome, type I.
- Вагнера синдром – характеризується наявністю очної симптоматики, подібної до синдрому Стіклера, при відсутності пораження суглобів, лицевих аномалій.
- Біндера синдром (максілоназальна дисплазія) характеризується гіпоплазією середньої частини обличчя, відсутністю передньої частини носової перетинки на Rо-графії.
- 50 відсотків пацієнтів з синдромом Стіклера мають П’єра Робена синдром.
- Кноблоха синдром характеризується високою міопією, енцефалоцеле частіше в потиличній ділянці.
- Ахондрогенез тип II – відсутність осифікації хребта, лобкових кісток; в/у смерть плоду або смерть в ранньому неонатальному періоді; всі випадки нові мутації.
- Гіпохондрогенез – помірний варіант ахондрогенезу.
- Вроджена спонділоепіфізарна дисплазія (SED congenita) – скелетні зміни більш виражені; низький зріст за рахунок тулуба; плоске обличчя, короткозорість.
- Спонділоепіметафізарна дисплазія (SEMD) Струдвіка тип – короткий зріст, кілевидна грудна клітка, сколіоз, розщелини піднебіння, відшарування сітківки; рентгенологічно нерегулярні склеротичні зміни метафізів довгих кісток, викликані чергуванням зон остеосклерозу та остеопенії, схожі з вродженою спонділоепіфізарною дисплазією.
- Хвороба Кніста – непропорційна карликовість; плоске обличчя, міопія, розщелини піднебіння, кіфосколіоз.
Stickler syndrome, type IІ.
- Маршалла синдром – гіпертелоризм очей; мікрогенія; маленький ніс; плоске обличчя, яке зберігається в дорослому віці; рентгенологічно гіпоплазія носових пазух, міопія, прогресуюча нейросенсорна глухота, П’єра Робена синдром, низький зріст, ранній артрит, гіпотрихоз, гіпогідроз.
Stickler syndrome, type ІІІ.
- Отоспонділометаепіфізарна дисплазія аутосомно-рецесивний тип спадкування (OSMED) – плоске обличчя, розщелини піднебіння, значна втрата слуху.
- Вайсенбаха-Цваймюллера синдром – неонатальний Стіклер синдром, характеризується гіпоплазією середньої частини обличчя, міопією, плоским переніссям, коротким носом, розщелинами хребта, мікрогнатією, нейросенсорним зниженням слуху, різомелічним вкороченням кінцівок, скелетні зміни зменшуються з віком, після 2-3 років ріст відповідає вікові.
Лікування:
- корекція розщелин піднебіння.
- корекція порушень зору;
- корекція порушень слуху;
- ортодонтична корекція щелепно-лицевих аномалій.
Література:
- About Stickler Syndrome. The Stickler Syndrome Support Group (SSSG) – https://stickler.org.uk/about-stickler-syndrome/.
- Stickler Syndrome. GeneReviews site.
- Stickler Syndrome, Type I; STL1. OMIM #108300.
- Stickler Syndrome, Type II; STL2. OMIM #604841.
- Stickler Syndrome, Type III; STL3. OMIM #184840.
Переглянуто редакційною колегією I.B.I.S.: 28/08/2003
Дивіться також:
Статевий розвиток хлопчиків
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”
Послідовність статевого розвитку –
середньостатистичний американський чоловік
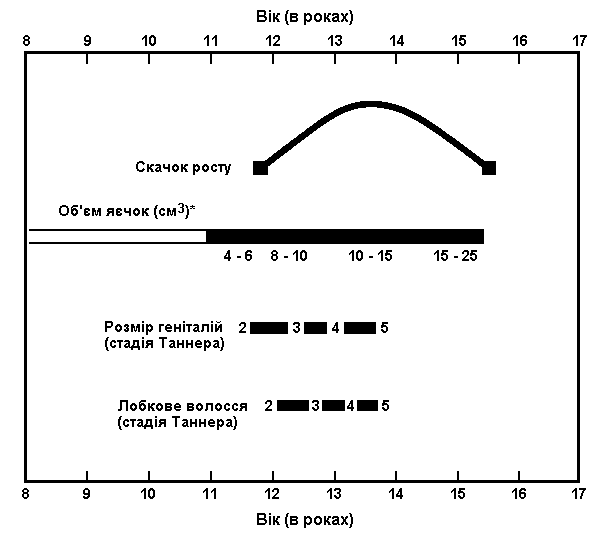
Адаптовано з Brookman et al: Princeton maturation study, 1976 (використовується з дозволу, Ross Laboratories).
|
|
|
|
|
|





