
Serhiy
Синдром Неймеген
(Nijmegen breakage syndrome)
С.О. Калинка
Лікар-генетик
Волинського обласного територіального медичного об’єднання захисту материнства і дитинства
Синоніми:
- Seemanova Syndrome II;
- Berlin Breakage Syndrome (BBS);
- Ataxia-Telangiectasia Variant V1; AT-V1.
Основні діагностичні критерії:
- пренатальна гіпоплазія;
- затримка фізичного розвитку;
- прогресуюча мікроцефалія з втратою когнітивних навичок;
- недостатність функції яєчників;
- часті респіраторні інфекції;
- підвищена чутливість до рентгенівського випромінювання;
- підвищений ризик злоякісних захворювань особливо лімфом;
- комбінований Т- і В-імунодефіцит;
- хромосомна нестабільність.
Клінічна характеристика:
Голова і шия: мікроцефалія зустрічається в 75% пацієнтів з народження, в решти на протязі перших місяців життя. Виступаюча середня частина обличчя, великий ніс, скошений лоб, гіпоплазія нижньої щелепи.
Вуха: великі вуха з диспластичними завитками.
Очі: монголоїдний розріз очей,телеангіектазії кон’юнктиви, епікант, пігментні вкраплення на очному дні.
Кисті і стопи: клинодактилія, полідактилія, широкий проміжок між першим і другим пальцями стопи, синдактилія стоп.
Шкіра: “кавові” плями, ділянки гіпер- і гіпопігментацій.
Респіраторна система: часті пневмонії, бронхіти, отити, синусити, мастоїдити, бронхоектази.
Урогенітальна система: аномалії нирок, атрезія прямої кишки.
Зріст і розвиток:
Пренатальна гіпоплазія, відставання у фізичному розвитку і затримка розумового розвитку зустрічається в більшості пацієнтів, але не в усіх випадках.
Відставання в фізичному розвитку може бути присутнім з народження, але стає очевидним в 2-річному віці. В ранньому дитинстві в більшості дітей погіршуються інтелектуальні здібності і після 7 років діагностують помірну розумову відсталість.
Первинна недостатність яєчників:
Первинна недостатність функції яєчників з підвищеним рівнем гонадотропіну, первинною аменореєю, затримкою статевого розвитку зустрічається у більшості осіб жіночої статі в препубертатному і пубертатному віці.
Підвищений ризик малігнізації:
Це найчастіше лімфоми: В–клітинні лімфоми, Т–клітинні лімфоми. Рідко зустрічаються гліоми, рабдоміосаркоми, медулобластоми.
Поєднанні вади розвитку:
Преаксилярна полідактилія, розщілини губи і піднебіння, гіпоплазія трахеї, підковоподібні нирки, гідронефроз, гіпоспадія, атрезія прямої кишки, дисплазія кульшових суглобів.
Частота:
Ймовірні коливання в межах 1:100000 живонароджених. Синдром Неймеген з найбільшою частотою зустрічається в східній європейсько-слов’янській популяції. Частота носійства в цих популяціях є 1:155.
Діагноз:
- характерні клінічні ознаки прогресуючої мікроцефалії (27-31см у новонароджених) з втратою когнітивних навичок, відставанням в зрості, частими респіраторними інфекціями, підвищеним ризиком злоякісних захворювань, особливо лімфом;
- діагноз підтверджується молекулярно-генетичним аналізом: виявленням гомозиготного носійства варіанту 657dеl5 гена NBSI, локалізованого у довгому плечі 8 хромосоми в ділянці 8q-21 майже в 100% пацієнтів.
Етіологія: аутосомно-рецесивний тип успадкування.
Лабораторні обстеження:
Клітинний і гуморальний імунодефіцит: в 35% випадків знаходять агаммаглобулінемію, в 20% пацієнтів – дефіцит імуноглубулінна А. Дефіцити Ig G2 і Ig G4 зустрічаються частіше, навіть якщо рівень імуногобуліна G є в нормі. Найбільш частими дефектами клітинного імунітету є зниження СД3+ Т клітин і СД4+ Т клітин.
Хромосомна нестабільність: в 10-50% метафаз PHA–стимульованих лімфоцитів виявляють інверсії 7 хромосоми і транслокації 7 і 14 хромосом. Найчастіше залучаються ділянки : 7p13, 7q35, 14q11, 14q32, які є локусами рецепторів Т-клітин та важких ланцюгів імуноглобулінів.
Чутливість до радіації: клітини пацієнтів, які піддавалися дії іонізуючого опромінення і радіоміметиків in vitro, мають знижену здатність до формування колоній.
Молекулярна діагностика: Можливі 2 типи молекулярно-генетичної діагностики:
- Виявлення варіанту 657del5 гена NBSI, локалізованого у довгому плечу 8 хромосоми в ділянці 8q21; Розвиток синдрому Неймеген пов’язаний з фунціональним дефіцитом білка нібрину (р95), який виникає внаслідок гомозиготного носійства варіанта 657del5, та деяких рідкісних варіантів гена NВS1. Всі уражені особи з Польщі, Греції і України, тестовані на сьогодні, є гомозиготами варіанта 657del5 гена NВS1. 70% пацієнтів, обстежених в Північній Америці, є гомозиготи варіанта 657del5, 15% є гетерозиготи варіанта 657del5 і ще однієї рідкісної мутації, решта 15% є гомозиготами рідкісних мутацій.
- Генетичний аналіз поліморфізму ДНК батьків і дитини (зчеплення генів).
Диференціальний діагноз:
- Часті респіраторні інфекції, відставання в зрості зустрічаються в інших спадкових імунодефіцитах. При агаммаглобулінемії Брутона і важких комбінованих імунодефіцитах також визначають підвищену чутливість до радіації.
- Мікроцефалія, виступаюча середня частина обличчя, затримка розумового розвитку зустрічаються в синдромах Секкеля, Nijmegen breakage syndrome-like disorder.
- Раннє відставання в фізичному розвитку присутнє в захворюваннях, які викликаються дефіцитом гормону росту, дефіцитом тіреоїдних гормонів, при скелетних дизплазіях.
- Пацієнти з лімфомами, які виявлені в 3-річному віці повинні уважно обстежуватись, тому що традиційні дози опромінення, які використовують в радіотерапії можуть бути потенційно небезпечні для хворих з синдромом Неймеген (NBS1).
Ризик для членів сім’ї:
- Батьки дітей з синдромом Неймеген є гетерозиготними носіями варіанта 657del5 та деяких рідкісних варіантів гена NBSI . Є повідомлення про підвищений ризик злоякісних захворювань у батьків.
- Ризик синдрому Неймеген в потомстві оцінюється як для захворювання з аутосомно-рецесивним типом успадкування.
Пренатальна діагностика:
Біопсія ворсин хоріона в 9-11 тижнів гестації, амніоцентез в 16-17 тижнів.
Лікування:
Специфічна терапія не розроблена. Проводиться часткова корекція імунного статусу шляхом замісної терапії внутрішньовенними імуноглобулінами.
У випадку онкологічних ускладнень проводиться поліхіміотерапія за ВFМ-протоколом для лікування лімфом, адаптованим до умов України.
Номер з каталогу МІМ:
251260 Nijmegen breakage syndrome.
Література:
- Carney JP, Maser RS, Olivares H, Davis EM, Le Beau M, Yates JR 3rd, Hays L, Morgan WF, Petrini JH. The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: linkage of double-strand break repair to the cellular DNA damage response. Cell 1998;93(3):477-86.
- Chrzanowska KH, Kleijer WJ, Krajewska-Walasek M, Bialecka M, Gutkowska A, Goryluk-Kozakiewicz B, Michalkiewicz J, Stachowski J, Gregorek H, Lyson-Wojciechowska G, et al. Eleven Polish patients with microcephaly, immunodeficiency, and chromosomal instability: the Nijmegen breakage syndrome. Am J Med Genet 1995;57(3):462-71.
- Van der Burgt I, Chrzanowska KH, Smeets D, Weemaes C. Nijmegen breakage syndrome. J Med Genet 1996;33(2):153-6.
- Varon R, Vissinga C, Platzer M, Cerosaletti KM, Chrzanowska KH, Saar K, Beckmann G, Seemanova E, Cooper PR, Nowak NJ, Stumm M, Weemaes CM, Gatti RA, Wilson RK, Digweed M, Rosenthal A, Sperling K, Concannon P, Reis A. Nibrin, a novel DNA double-strand break repair protein, is mutated in Nijmegen breakage syndrome. Cell 1998;93(3):467-76.
- Wegner RD, Chrzanovska KH, Sperling K, Stumm M. Ataxia-Telangiectasia variants (Nijmegen breakage syndrome). In Ochs HD, Smith CIE, Puch JM. Primary Immunodeficiency Diseases, a molecular and genetic approach. Oxford University Press, Oxford UK. 1999:324-34.
- Синдром Ніймеген: Поради лікаря-генетика /Інститут спадкової патології АМН України. – Львів, 2 с.
Переглянуто редакційною колегією I.B.I.S.: 01/08/2024
Вимірювання руки
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”



Дані базуються на популяційному дослідженні 2403 дітей від народження до 14-річного віку. 56% становили діти чоловічої статі, 44% – жіночої; 2006 відносились до білої раси, 206 – до темношкірої, 43 – азійської. Feingold M та Bossert WH: Birth Defects: Orig Art Series X(13), 1974.
Підковоподібна нирка (ПН)
(Horseshoe Kidney)
І.Т. Мороз
Лікар-неонатолог відділення патології новонароджених
Рівненської обласної дитячої лікарні
Включені:
Вади розвитку, пов’язані з порушенням співвідношення метанефрогенних бластем (вади взаємного розміщення нирок).
Етіопатогенез:
Зрощення нирок виникає внаслідок ненормального злиття парних метанефрогенних бластем, яке проходить на ранніх етапах диференціації. Цей процес відбувається до того, як нирки піднімуться над біфуркацією аорти – це відповідає приблизно 8-10 тижневі ембріонального розвитку.
Вада виникає внаслідок двох факторів: ненормальне злиття метанефрогенних бластем, зумовленого цим, відхилення від нормального ходу ембріофетальної міграції нирок.
Вростання протоків метанефросів у метанефрогенні бластеми та індуковані ними диференціації метанефрогенної тканини в кінцеву ниркову тканину відбуваються без суттєвих відхилень, гістологічно від норми не відрізняється.
При збереженні монолатерального розміщення нирки можуть зростатись полюсами (верхнім або нижнім).
Морфологічні форми:
У 90% випадків зрощення спостерігається в зоні нижніх полюсів.
- Зрощення нижніх полюсів за рахунок паренхіматозного, достатньо масивного перешийка. Перешийок розміщений над аортою і нижньою порожнистою веною, на рівні ІІІ-IV поперекових хребців або нижче.
- Безпосереднє зрощення нижніх полюсів всією їх масою.
- Паренхіматозний перешийок може з’єднати зони нирок дещо вище їхніх нижніх полюсів. При цьому нижні полюси можуть бути вільними.
- Перешийок між нижніми полюсами може бути утворений фіброзною тканиною, яка позбавлена або майже позбавлена елементів ниркової паренхіми.
- Перешийок (частіше фіброзний) може розміщуватись під аортою і нижньою порожнистою веною або між ними (10% випадків цієї вади).
Нирки можуть зростися верхніми полюсами, утворюючи підкову відкриту донизу. Половини підковоподібної нирки неоднакові за розмірами, часто мають дольчасту будову, іноді нерівномірно деформовані. Одна з половин може бути недорозвиненою. Гістологічно будова підковоподібної нирки не відрізняється від будови нормальної нирки.
За будовою мисково-чашкової системи на основі розміщення мисок розрізняють три типи підковоподібної нирки:
- Найбільш поширений – обидві миски розміщені на передній поверхні зрощених нирок.
- Миски розміщені внутрішньонирково, але також на передній поверхні нирок.
- Одна або обидві миски розміщені на внутрішній або навіть на задній поверхні зрощених нирок.
Незвична також будова ниркових чашок. Частіше верхня група розвинута більше, ніж нижня. Шийки верхніх чашок витягнуті і відхилені до середини. Верхні чашки своїми форнікальними зонами здебільшого звернені в латеральну зону. Нижні чашки недорозвинені, зміщені до середньої лінії і часто звернені своїми форніксами в медіальну сторону.
Підковоподібна нирка відображається на будові верхніх відділів сечоводів. Навколомискові відділи сечоводів зазвичай відхилені латерально, а потім конвергують в напрямку до середньої лінії. Обидва сечоводи, як правило, вкорочені. Можливе стиснення сечоводу перешийком підковоподібної нирки з розвитком внаслідок цього гідронефрозу.
Клініка та клінічне значення:
Проблема набуває важливості по мірі того, як підковоподібна нирка сама по собі стає джерелом клінічних проявів, в якій мірі схильна до ускладнень, захворювань.
Внаслідок особливостей кровопостачання, інервації і тиску перешийка на нервові сплетіння і у здоровій підковоподібній нирці може виникнути характерний больовий симптомокомплекс. Аномальна нирка нерідко супроводжується біллю в ділянці пупка під час розгинання тулуба або перегинання назад (симптом I. Rovsing). Біль тупий, може бути інтермітуючий або постійний. Стиснення аорти спостерігається рідко, та якщо є, то супроводжується симптомами коарктації аорти.
Стиснення нижньої порожнистої вени перешийком ПН може призвести до венозної гіпертензії нижньої половини тіла з наступною дилатацією і венозним застоєм в цій ділянці. Це в свою чергу може обумовити набряк нижніх кінцівок і навіть асцит. Іноді з’являється відчуття оніміння в нижніх кінцівках.
Підковоподібна нирка являє собою значне відхилення від норми і не може не проявлятись клінічно: описана періодична макрогематурія, яка виникає при здоровій підковоподібній нирці внаслідок внутрішньониркової гіпертензії.
ПН схильна до виникнення гідронефрозу, специфічних і неспецифічних захворювань, нефрогенної артеріальної гіпертензії.
Найчастішими ускладненнями є пієлонефрит – 45%, гідронефроз – 26%, нефролітіаз – 22%.
Діагностика:
- Фізикальні методи: при прощупуванні органів черевної порожнини у 10-15% осіб, що мають ПН, пальпується її перешийок у вигляді щільного довгастого або округлого нерухомого утвору, що розміщується в мезогастральній ділянці. Прощупування перешийка може бути чутливе.
- Рентгенологічні методи: характерна тінь зрощених нирок іноді може бути виявленою на оглядовій урограмі. Екскреторна урографія і ретроградна пієлоуретрографія дають достатньо інформації про наявність ПН, особливості будови її миско – чашкової системи і сечоводів. У процесі урографічних досліджень розпізнаються захворювання ПН (калькульоз, гідронефроз). У дітей обов’язкова цистографія, оскільки висока ймовірність міхурово – сечоводних рефлюксів.
- Радіоізотопні методи дослідження: часто спостерігаються накопичення ізотопа в перешийку, що допомагає відрізнити паренхіматозний перешийок від фіброзного.
- Ангіографія: вивчення ангіоархітектоніки, що дозволяє встановити не тільки кількість артерій, але й число, локалізацію додаткових судин, дає можливість визначити товщину, особливості кровопостачання перешийка, наявність функціонуючої паренхіми, відношення перешийка до великих судин, що є важливим при оперативному втручанні.
- УЗД: розрізняють перешийок ПН в тих випадках, коли в ньому слабка васкуляризація, внаслідок чого він погано диференціюється при радіоізотопних і ангіологічних дослідженнях.
Асоційовані вади:
ПН часто поєднується з іншими вадами розвитку, і може бути складовою частиною багатосистемних вад розвитку, які відносяться до складу численних спадкових синдромів та хромосомних захворювань, а саме:
- При порушенні в системі аутосом – трисомія Е, трисомія G, делеція 5р, делеція 18q.
- Синдром Шерешевського-Тернера (45ХО, 45ХО/46ХХ).
- Синдром Патау (трисомія 13-15).
- Синдром Бартолін-Патау – тип успадкування – домінантний, зчеплений із Х хромосомою.
- Синдром Ландольфа (синдром ТАР) – тип успадкування аутосомно-рецесивний.
- Синдром Гутіреза – тип успадкування не встановлено.
Звертає на себе увагу те, що в описаних спадкових синдромах досить часто зустрічаються вади розвитку опорно – рухового апарату, щелепно – лицьової ділянки і аналізаторів (слухового, зорового), які повинні розцінюватись як стигми дизембріогенезу органів сечостатевої системи. Тому при виявленні вказаних вад розвитку вкрай необхідні активні, із застосуванням сучасних методів, дослідження, пошуки вад розвитку нирок і сечоводів. Оскільки тип успадкування стосовно більшості багатосистемних вад розвитку встановлено досить точно, створюється можливість активного виявлення вад розвитку нирок і сечоводів в їх доклінічній стадії серед членів родини пробанду.
Частота:
Складає 1:425 – 1:700 випадків, які виявлено при секції.
Лікування:
При здоровій ПН оперативне втручання, направлене на створення анатомічних умов, наближених до нормальних, істмотомія в даний час доцільна. Больовий синдром при ПН надзвичайно рідко обумовлюється тільки пересіченням хребта перешийком. Тому пересічення перешийка і розсунення зрослих полюсів латерально не приносить полегшення хворому. Істмотомія виправдана в тих випадках, коли перешийок, стиснувши аорту або порожнисту вену, є причиною венозного стазу в нижній половині тулуба і порушує кровопостачання окремих органів.
Резекція перешийка показана при гідронефрозі, рецидиві сечокам’яної хвороби і інших патологічних станів.
Номер з каталогу МІМ:
602200 Ventriculomegaly With Defects of the Radius and Kidney.
Література:
- Айвазян А.В., Войно-Ясенецкий А.М. Пороки развития почек и мочеточечников.- М.: Наука, 1988.- C. 87-186.
- Лопаткин Н.А., Люлько А.В. Аномалии мочеполовой системы.- К.: Здоров’я, 1987.- С. 80-91.
- Warkany J. Congenital malformations. Notes and comments. Chicago 1981:1050.
Переглянуто редакційною колегією I.B.I.S.: 05/02/2002
Нейрофіброматоз
(Neurofibromatosis)
Наталія Афанасьєва
Завідуюча Кримським республіканським медико-генетичним центром,
кандидат медичних наук
Нейрофіброматоз відноситься до групи факоматозів (грецькою phakos – пляма, matosis – пухлина). Сьогодні ця назва поєднує кілька захворювань, які насправді є різними нозологічними формами: нейрофіброматоз I, II, III і IV типів. Біля 90% усіх хворих на нейрофіброматоз мають нейрофіброматоз I типу.
Нейрофіброматоз I типу (НФ1, класичний, периферійний, власне хвороба Реклінгхаузена, номер в OMIM – 162200) — це системне спадкове захворювання з переважним ураженням шкіри та нервової системи, успадковується аутосомно-домінантно, з високою пенетрантністю і варіабельною експресивністю. Захворювання обумовлене мутацією гену “NF1” в 17q-хромосомі. Чоловіки та жінки уражуються однаково часто.
Нейрофіброматоз був описаний німецьким патологом Фрідріхом Даніелем фон Реклінгхаузеном (Recklinghausen F.D.) в 1882 році в роботі “Про множинні фіброми шкіри та їх зв’язок з множинними невромами”, після цього захворювання отримало його ім’я.
НФ1 – це одне з найпоширеніших моногенних захворювань людини, зустрічається з частотою не рідше 1:3000 — 1:4000 населення. Загальна кількість хворих у світі наближається до 1 мільйону. В Україні кількість хворих становить приблизно 10-11 тисяч (достовірні дані про частоту цього захворювання в нашій країні відсутні).
Етіологія та патогенез:
Ген NF1 локалізується на довгому плечі 17 хромосоми (17q11.2). Приблизно половина випадків — це наслідок нових мутацій. Частота мутацій гену NF1 визначена як 1 х 1014 (одна з найвищих серед спадкових захворювань людини). При цьому захворюванні описані різні типи мутацій: великі та малі делеції, транслокації гену, заміни нуклеотидів (всього більше 100 варіантів), що значно ускладнює їх пошук під час молекулярно-генетичного дослідження.
НФ1 має повну (100%) пенетрантність, тобто усі носії патологічного гену є хворі, але експресія гену, тобто ступінь викликаних ним клінічних проявів, дуже варіюється. Навіть в межах однієї сім’ї можуть спостерігатися як мінімальні прояви, так і важкі випадки.
Ген NF1 є одним з основних генів-супресорів пухлинного росту для 30% тканин організму, в першу чергу нейроектодермального походження. Продуктом гену NF1 є великий білок нейрофібромін, який забезпечує контроль за ростом клітин. При пошкодженні цього гену в одній з хромосом 17-ої пари 50% нейрофіброміну, що синтезується, стає дефектним і спостерігається зміщення рівноваги росту клітин в бік проліферації та/або недостатньої диференціації клітин, що зрештою призводить до розвитку пухлин.
Захворювання має виражений клінічний поліморфізм, прогресуючий перебіг, поліорганність уражень та високу частоту ускладнень (злоякісне переродження нейрофібром, судомні напади тощо).
Діагностичні критерії НФ1:
Клінічна діагностика НФ1 ґрунтується на виявленні діагностичних критеріїв, рекомендованих Міжнародним комітетом експертів з нейрофіброматозу при Національному інституті здоров’я США в 1987 році. Діагноз може бути поставлений при наявності у хворого не менше двох з перелічених нижче ознак:
- Не менше п’яти плям кольору “кави з молоком” діаметром більше 5 мм у дітей препубертатного віку і не менше шести таких плям діаметром більше 15 мм в постпубертатному віці.
- Дві та більше нейрофіброми будь-якого типу чи одна плексиформна нейрофіброма.
- Множинні дрібні пігментні плями по типу веснянок, які локалізуються у великих складках шкіри (під пахвами та/або в паху) – симптом Кроува (Crowe).
- Гліома зорового нерву.
- Два та більше вузликів Ліша на райдужній оболонці, їх можна побачити при огляді за допомогою щілинної лампи.
- Дисплазія крила клиноподібної кістки або вроджене потоншання кортикального шару довгих трубчастих кісток з наявністю псевдоартрозу чи без нього.
- Наявність у родичів першого ступеня нейрофіброматозу I типу за тими ж критеріями.
Особливістю захворювання є специфічна послідовність проявів симптомів в залежності від віку пацієнта, що ускладнює клінічну діагностику НФ1 в ранньому дитячому віці. Таким чином, від народження та протягом перших років життя можуть існувати лише деякі ознаки нейрофіброматозу I типу, наприклад, великі пігментні плями, плексиформні нейрофіброми, скелетні дисплазії. Інші симптоми можуть проявлятися значно пізніше (в 5–15 років). При цьому виразність клінічних проявів, протікання та швидкість прогресування НФ1 у різних хворих неоднакові і коливаються в широких межах.
Найчастіше трапляються прояви з боку шкіри. Пігментні плями чітко окреслені, світло-коричневого кольору, можуть зустрічатися при народженні, проте найчастіше з’являються протягом першого року життя (у 82% випадків). До 4-го року життя (або раніше) вони реєструються в усіх дітей з НФ1 (фото 1).
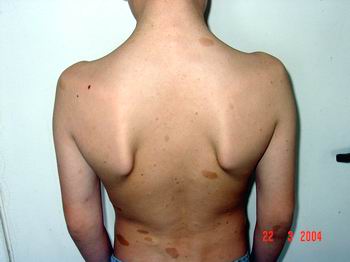
Нейрофіброми (дермальні, гіподермальні, плексиформні) є найвиразнішим проявом хвороби Реклінгхаузена, їх кількість іноді сягає кількох сотень (фото 2).
Нейрофіброми при народженні зустрічаються рідко, частіше вони з’являються у 3-5 років. Час від часу їх кількість і розміри зростають внаслідок дії різних стимулюючих факторів, серед яких найважливішими є гормональна перебудова організму: пубертатний вік, період вагітності та після пологів, а також перенесені травми або тяжкі соматичні захворювання. Із збільшенням спектру пропонованих комерційних медичних та косметичних послуг значно збільшилась кількість звернень хворих, які вказують на появу нових пухлин (нейрофібром, неврином, шванном) після ятрогенних втручань. Мова йде про видалення пухлин з діагностичною або лікувальною метою за допомогою різних методів, включаючи хірургічне вирізання. До ятрогенних ускладнень також призводить призначення фізіотерапевтичних процедур при лікуванні різних соматичних захворювань, корекція скелетних порушень (різноманітних видів сколіозу, переломів) та нервово-м’язових розладів (дуже часто дітям грудного віку призначається масаж, коли діагностика нейрофіброматозу I типа неможлива через недостатність клінічних проявів). Але часто захворювання прогресує і на фоні гаданого благополуччя.
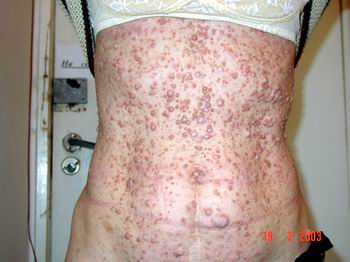
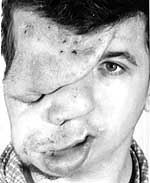
Плексиформна нейрофіброма являє собою м’яку рихлу масу, яка „повзе” по ходу нерва, як правило, трійчастого або верхнього цервікального, іноді з інтенсивним коричневим забарвленням, і яка є значним косметичним дефектом (фото 3). Плексиформні нейрофіброми можуть бути гігантськими, масою більше 10 кг, часто вони пов’язуються з підвищеним ризиком переродження у злоякісні пухлини. При локалізації в середостінні, в черевній порожнині, в очній ямці вони призводять до порушення функцій прилеглих органів. Зустрічаються приблизно у 4% хворих.
Гістологічно нейрофіброми складаються з фібробластів, клітин Шванна, периневральних клітин. Крім того, в складі нейрофібром є велика кількість тучних клітин. При електронній мікроскопії спостерігається їх тісний контакт з лімфоцитами.
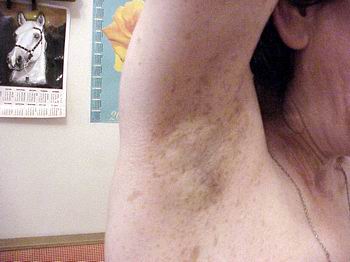
Симптом Кроува (Crowe) — дифузна пігментація (по типу веснянок) під пахвами або в крупних складках (фото 4). Симптом зустрічається у 70% хворих на НФ1, як правило, починаючи з середнього дитячого віку.
Вузлики Ліша – це пігментована меланоцитна гамартома райдужної оболонки, яку знаходять при огляді за допомогою щілинної лампи. У випадку множинних вузликів цей симптом є патогномонічний для НФ1 і спостерігається у більш ніж 90% хворих протягом другого десятиріччя життя.
Супутні порушення інших органів та систем:
З боку опорно-рухового апарату часто бувають сколіози, кіфози, деформації грудної клітки. Остеопороз зустрічається у більше ніж 90% випадків. Витончення кортикального шару довгих трубчастих кісток може призводити до формування несправжніх суглобів.
Найхарактернішими психічними порушеннями є розумова відсталість, затримка мовного розвитку, дислексія, когнітивні порушення, які спостерігаються у 20-30% дітей. Інтелектуальна недостатність звичайно пов’язана з недостатністю пам’яті, уваги, динаміки психічних процесів.
Основним завданням наукових досліджень є розробка методів патогенетичного лікування нейрофіброматозу I типу, які б дали змогу стримувати появу нових і ріст існуючих пухлин, а також запобігати розвитку ускладнень.
Нейрофіброматоз II типу (НФ2):
НФ2 (центральний нейрофіброматоз, двобічний слуховий нейрофіброматоз, код по OMIM – 101000) – це прогресуюче інвалідизуюче моногенне аутосомно-домінантне захворювання, яке проявляється двобічними вестибулярними шванномами, а також множинними пухлинами центральної та периферичної нервової системи, ранніми катарактами. Зустрічається з частотою 1:200000 населення.
Міжнародним комітетом експертів з нейрофіброматозу при Національному інституті здоров’я США в 1987 році рекомендовані діагностичні критерії НФ2:
- Двобічна невринома слухового нерву, підтверджена МРТ.
- Наявність НФ2 у прямих родичів.
- Однобічна пухлина (новоутворення) VIII черепного нерву або
- Наявність двох з наступних ознак: нейрофіброма, менінгіома, гліома, шваннома, ювенільна задня субкапсулярна катаракта.
Ген НФ2 знаходиться в 22 хромосомі (22q12). Продукт гену – білок мерлін (шванномін) є білком-супресором пухлин. Мерлін по структурі та властивостях дуже близький до трьох гомологічних білків: моезіну, езріну і радіксіну. Найбільше значення вони мають в регулюванні проліферації клітин нейроектодермального походження. Детально патогенез захворювання на сьогоднішній день не досліджений, однак багато авторів вказують на схожі ланки етіопатогенезу при НФ2 і спорадичних менінгіомах та шванномах.
Симптоми НФ2 звичайно з’являються під час другого десятиріччя життя або пізніше.
Двобічна невринома слухових нервів реєструється у більше ніж 90% хворих. Захворювання маніфестує, як правило, шумом у вухах, зниженням слуху. Крім того, зустрічаються менінгіоми, шванноми, епендімоми головного та спинного мозку, черепно-мозкових нервів. При цьому з’являються скарги на порушення координації, ністагм, головні болі. Можуть розвиватися судомні напади.
Плями кольору “кави з молоком” і периферичні нейрофіброми зустрічаються також у хворих на НФ2, але рідше, ніж при НФ1. Звичайно їх буває менше 6.
Нейрофіброматоз III типу:
Нейрофіброматоз III типу (код по OMIM – 162260) – долонні нейрофіброми, бліді відносно великі плями кольору кави з молоком, двобічні невроми слухового нерву, менінгіоми задньої ямки та верхнього шийного відділу, спінальні та параспінальні нейрофіброми; відсутність вузликів Ліша на райдужці; пухлини центральної нервової системи, що швидко розвиваються під час другого і третього десятиріччя життя.
Нейрофіброматоз IV типу:
Нейрофіброматоз IV типу (код по OMIM – 162270) – клініка нейрофіброматозу I типу, але без вузликів Ліша.
Лікування:
Радикального методу лікування нейрофіброматозу поки що не існує. Вчені сконцентрувались на можливості етіологічного лікування, тобто генної інженерії. Особливо далеко цей напрямок просунувся з моменту відкриття мутантного гену та розшифровки його первинного продукту — нейрофіброміну в 1990 році. На наукові дослідження, пов’язані з цією проблемою, щорічно виділяються величезні кошти, однак результатів, які можна було впровадити в практичну медицину, поки не досягнуто.
Для лікування цього захворювання традиційно використовуються методи симптоматичної терапії: хірургічне видалення пухлин або променева терапія нейрофібром внутрішніх органів. Проте ці методи не можна використовувати в якості методів вибору, враховуючи множинність пухлин, а також можливість рецидиву та прискорення росту інших нейрофібром після хірургічного втручання. Це ж можна сказати й про використання лазерної хірургічної методики видалення усіх нейрофібром на поверхні шкіри та в підшкірному шарі хворого, тому що це не рятує від виникнення нових чи посиленого росту існуючих гамартом в центральній нервовій системі. Вочевидь, хірургічні методи лікування нейрофіброматозу повинні застосовуватись за суворими показами: болі, порушення чутливості та рухових функцій, швидкий ріст пухлини, з обґрунтуванням термінів операції.
Перша спроба патогенетичного підходу до лікування була зроблена V. Riccardi в 1987 році, коли він запропонував довготривале використання кетотіфену (в дозі 2-4 мг протягом 1,5-3 років) для стабілізації мембран тучних клітин, вважаючи, що саме дегрануляція цих клітин стимулює ріст пухлин. Проте лікування одним кетотіфеном не принесло очікуваних результатів: зменшувались суб’єктивні відчуття болю і свербіння в ділянці нейрофібром, але якого-небудь впливу на ріст пухлин відмічено не було.
З того часу багато вітчизняних і закордонних авторів пропонували свої методи медикаментозної терапії, направленої на запобігання появі нових нейрофібром і сповільнення зростання існуючих. Так, в Центральному науково-дослідному шкірно-венерологічному інституті (Росія) у відділі спадкових захворювань шкіри розроблена методика лікування хворих на нейрофіброматоз, яка включає препарати, що гальмують дегрануляцію тучних клітин (кетотіфен та його аналоги), знижують рівень в тканинах глікозамінгліканів (лідаза та її аналоги), засоби з антипроліферативним ефектом (тігазон, вітамін А у великих дозах). Аналогічні схеми лікування пропонуються й іншими авторами з ширшим використанням системних ензимних препаратів, препаратів-інгібіторів синтезу ДНК, інгібіторів синтезу цитокінів та інш. Вищевказані препарати використовуються комплексно в різних комбінаціях або у вигляді монотерапії залежно від форми нейрофіброматозу, скарг, перебігу, а також віку і статі хворих. Обов’язково лікування проводилось під час прогресування захворювання, тобто при появі нових пухлин та/або рості існуючих, які, як правило, супроводжуються свербінням або відчуттям болю в їх проекції, а також з метою запобігання активізації хвороби під час запланованих операцій на пухлинах.
Нижче наведено рекомендації Американської академії педіатрії щодо медичного нагляду за дітьми з нейрофіброматозом (pdf-файл).
Медичний нагляд за дітьми з нейрофіброматозом
(Схвалено Американською педіатричною академією)
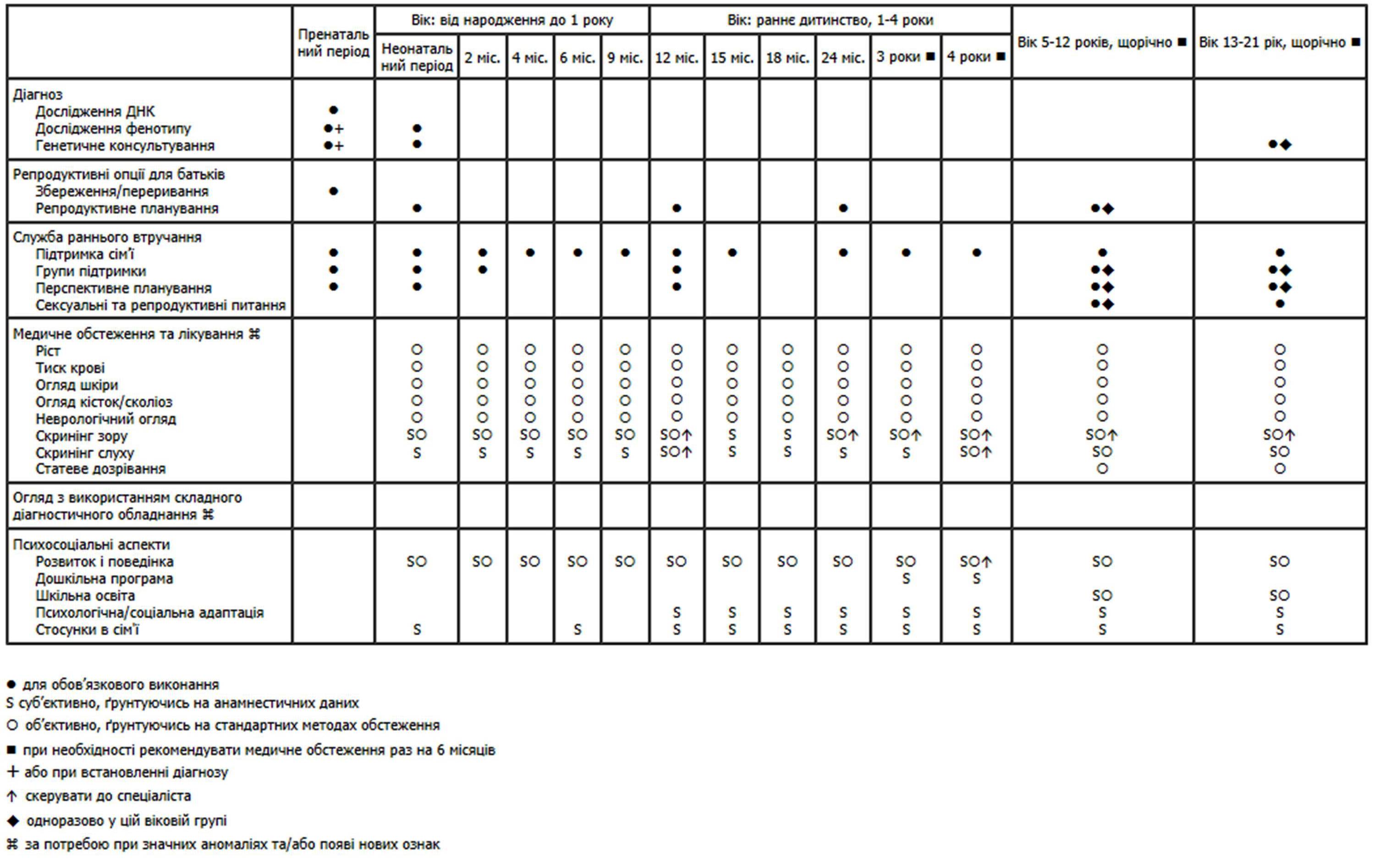
Література:
- Каталинич Д. /Хирургическое лечение нейрофиброматоза с помощью лазера. //Хирургия. – № 5. – 1996.
- Козлов А.В. /Наследственные заболевания в нейроонкологии. //Вопросы нейрохирургии. – № 3. – 2003.
- Короленко В.В., Бондур В.В. /Фармакотерапія нейрофіброматозу I типу як альтернативний метод лікування: сучасні досягнення і перспективи розвитку. //Дерматологія та венерологія. – № 4(18). – 2002.
- Макурдумян Л.А. /Нейрофиброматоз I типа. Проблемы диагностики и лечения. //Лечащий врач. – № 10. – 2001.
- Михайловский М.В. /Деформации позвоночника при нейрофиброматозе: обзор литературы. //Хирургия позвоночника. – № 3. – 2005.
- Мордовцева В.В., Мордовцева В.В. /Нейрофиброматоз I типа (болезнь Реклингаузена): ген открыт, клинико-патогенетические и терапевтические проблемы остаются. //Российский журнал кожных и венерических болезней. – № 2. – 1999.
- Цимбалюк В.І, Квасніцький М.В. /Діагностика та лікування нейрофіброматозу. //Мистецтво лікування. – № 5 (011). – 2004.
- Health Supervision for Children With Neurofibromatosis. American Academy of Pediatrics policy (http://aappolicy.aappublications.org).
- Packer RJ, Rosser T. Therapy for plexiform neurofibromas in children with neurofibromatosis 1: an overview. J Child Neurol. 2002 Aug;17(8):638-41.
Переглянуто редакційною колегією I.B.I.S.: 15/11/2005
Дивіться також:
- Нейрофіброматоз (інформація для батьків)
Нейрофіброматоз
(Neurofibromatosis)
Сергій Лапченко
Інформаційний спеціаліст Волинського ОМНІ-Центру
Тетяна Сороцька
Інформаційний спеціаліст Рівненського ОМНІ-Центру
Нейрофіброматоз (НФ) – це сукупність спадкових розладів нервової системи. На сьогоднішній день відомі дві його форми: НФ1 (периферичний нейрофіброматоз, хвороба Реклінгаузена) та НФ2 (білатеральний акустичний нейрофіброматоз, центральний нейрофіброматоз). У розвинутих країнах (наприклад, у США) більш розповсюдженим є НФ1, на нього хворіє одна дитина з кожних 4000. НФ2 є менш розповсюдженим і трапляється 1 на кожні 40000 народжень.
Ступінь проявів хвороби не залежить від форми нейрофіброматозу, вона змінюється від дуже сильних до часом майже непомітних симптомів.
Раніше користувались кількома іншими назвами цього захворювання, але в результаті останніх змін в розумінні його природи, вони виявились недоречними. НФ1 називався периферійним нейрофіброматозом або хворобою Реклінгуазена на честь лікаря, який першим описав його у 1982 році. Для НФ2 використовували терміни “двобічний акустичний нейрофіброматоз, центральний фіброматоз або вестибулярна шваннома”.
Нейрофіброматоз не залежить від статі, расової чи етнічної групи. Відомо, що його причиною є дефекти певних генів. Тому нейрофіброматозом не можна заразитися від вже хворої людини, як це трапляється, наприклад, з туберкульозом або грипом.
Певний час вважали, що деформація тіла в головного героя книжки, фільму і вистави “Людина-слон” викликана НФ. Останні дослідження вказують на те, що це результат синдрому Протея. Хворим на НФ не варто хвилюватись, що вони матимуть фігуру, як в Людини-слона.
Які основні прояви нейрофіброматозу?
Звичайно, раннім проявом НФ1 є шість або більше великих коричневих плям на шкірі. Лікарі називають їх плямами кольору “cafе-au-lait”, що французькою мовою означає “кава з молоком”. Часто ці плями можна помітити відразу після народження і з часом їх розмір і кількість збільшуються, а колір може темнішати. До 2 років життя більше половини хворих дітей мають такі плями.
Іншим проявом НФ1 є маленькі доброякісні пухлини у вигляді бугорків під шкірою або глибше. Вони можуть проявитися в любому віці, але найчастіше в юнацькому періоді. Ці пухлини ростуть з нервів і складаються з нервових та інших клітин, тому їх називають нейрофібромами. Хворий може мати сотні таких нейрофібром, але треба пам’ятати, що часом буває нейрофіброма у людини, яка не є хворою на НФ1.
Часом пухлини ростуть на зоровому нерві і, в такому випадку, можуть суттєво впливати на зір. Маленькі вузлики (пухлинки) можуть з’явитися на райдужці (розфарбована частина ока). Вони, звичайно, проявляються після досягнення статевого віку, не спричиняють ніяких неприємностей, але можуть допомогти лікарю діагностувати нейрофіброматоз, тому що у дорослих вони можуть бути єдиним симптомом НФ1.
НФ2 характеризується пухлинами, які ростуть на 8-му черепному нерві (один із 12 пар нервів, що ідуть до головного мозку). Ці пухлини часто затискають слуховий нерв, викликаючи глухоту. Пухлини також можуть рости і в інших частинах головного і спинного мозку, часом у хворих дітей в ранньому віці розвиваються катаракти. Коричневі плями і пухлини під шкірою не характерні для НФ2, їх або немає зовсім, або буває дуже мало. Симптоми НФ2 звичайно проявляються лише під час статевого дозрівання (лікарі називають цей час пубертатним періодом).
Що викликає нейрофіброматоз?
Причиною виникнення як НФ1, так і НФ2 є певні аномальні гени, які або успадковуються від когось із хворих батьків, або з’являються самостійно внаслідок нової мутації (зміни структури) в гені. Таким чином, нейрофіброматоз може з’явитись в сім’ї, історія якої ніколи не знала такої хвороби. Приблизно 50% випадків нейрофіброматозу спричинені новою мутацією генів. Симптоми хвороби, викликаної мутацією, точнісінько такі ж, як і симптоми хвороби, успадкованої від батьків.
Ген успадковується по аутосомно-домінантному типу, тому дитина, один із батьків якої хворий на НФ, має 50% шансів успадкувати цю хворобу. Проте, в неї є і 50% шансів народитися здоровою, оскільки вона може дістати нормальні гени від здорового батька чи матері.
Як нейрофіброматоз впливає на життя хворого?
В більшості випадків симптоми НФ1 є помірні і хворі живуть нормальним життям. Часом хворий має велику кількість нейрофібром шкіри на обличчі або на тілі і вони можуть збільшуватись під час статевого дозрівання у зв’язку з гормональними змінами в організмі. Звичайним явищем при нейрофіброматозі є прогресуючий сколіоз (прогресуюче викривлення хребта).
Деякі діти, хворі на НФ1, мають напади, проблеми з навчанням, вони погано говорять і можуть бути гіперактивними, часом у них буває велика голова. Звичайно, це не викликає серйозних медичних проблем. Пухлини в оці чи у вусі можуть бути причиною випинання ока чи погіршення зору і слуху. При народженні часом виявляють різні вади розвитку кісток, включаючи деформацію ніг нижче коліна. Діти з НФ1 мають підвищений ризик захворіти на певні види раку, хоча це трапляється дуже рідко.
Серед симптомів НФ2 зустрічаються втрата слуху, дзвін у вухах, запаморочення, проблеми з підтриманням рівноваги, головні болі та напади. Вчасне видалення невеликих пухлин слухового нерву допомагає зберегти слух. Нова діагностична процедура, магніто-резонансна томографія, може виявити такі пухлини в ранньому періоді їх розвитку.
Як і у випадках з іншими генетичними хворобами, у пацієнтів з НФ можуть виникнути проблеми психологічного характеру. Деякі з них уникають спілкування, стають відчуженими, тому що почувають себе несхожими на інших. Вони можуть вагатись, чи їм слід мати дітей, хвилюватись про можливі ускладнення в майбутньому. На жаль, сьогодні неможливо передбачити ступінь проявів нейрофіброматозу у хворого.
Чи можливо вилікувати нейрофіброматоз?
Ліків від нейрофіброматозу немає, проте лікування його наслідків є можливим і навіть необхідним. Болючі і деформуючі тіло пухлини шкіри можуть бути видалені хірургічним шляхом, проте, на жаль, вони можуть з’являтися знову. Часом можна видалити пухлини на очному та слуховому нервах. Зі сколіозом можна боротися хірургічно або за допомогою спеціальних фізичних методів.
Є надія, що останні дослідження сприятимуть покращанню лікування НФ1 та НФ2. Вчені виявили розташування гену НФ1 на 17-ій хромосомі і гену НФ2 на 22-ій хромосомі. Це відкриття допоможе у розробці тесту з точними показниками для діагностики захворювання ще до прояву симптомів. Завдяки ранній діагностиці лікарі зможуть раніше розпочати лікування, і, таким чином, запобігти деяким серйозним ускладненням. Дослідники виявили також протеїни, що утворюються цими генами і наблизились до розкриття їх функцій. Результатом цього може стати розробка медикаментів для лікування деяких симптомів нейрофіброматозу.
У рідких випадках нейрофіброматоз не має ознак ні НФ1, ні НФ2, або, навпаки, має ознаки обох цих форм. Такі випадки вивчені дуже мало.
Чи можливо виявити нейрофіброматоз до народження?
Сьогодні тільки генетичне тестування і тільки в сім’ях, де вже були подібні випадки, може діагностувати або, в крайньому разі, вирахувати наявність НФ1 у плода.
Існують два основних види генетичного обстеження. Перший називається аналізом зчеплення, і використовується при обстеженні сімей, в яких є більше двох хворих. Для проведення аналізу зчеплення необхідні проби крові від багатьох членів родини. При цьому виді аналізу простежують хромосому, яка містить змінений ген – чинник хвороби, у представників двох або більше поколінь. Аналіз зчеплення показує ймовірність виникнення НФ з точністю понад 95%. Для другого виду генетичного обстеження – аналізу прямої генної мутації – непотрібне обстеження інших членів сім’ї. Але цей аналіз придатний не для всіх випадків, тому що він залишає невиявленими третину мутацій, які спричиняють НФ1 та НФ2. Очікується його вдосконалення у найближчі кілька років.
Генетичне консультування може допомогти тим людям, які хворі на нейрофіброматоз і планують мати дітей. Генетики пояснять ризики і альтернативи, дадуть відповіді на запитання, зважать необхідність проведення пренатальної діагностики і запропонують підтримку.
Переглянуто редакційною колегією I.B.I.S.: 05/02/2002
Дивіться також:
- Нейрофіброматоз (інформація для спеціалістів)
Дерматоми
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”
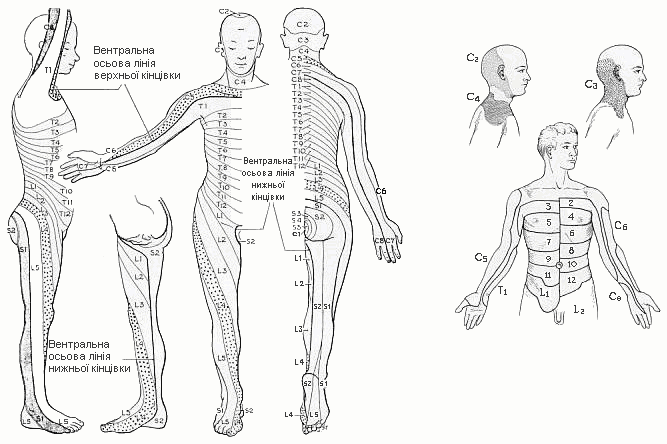
Дерматоми – сегментний розподіл шкірних нервів. Grant’s Atlas of Anatomy, ed.6, Williams and Wilkins, 1972.
Ретинопатія недоношених
(Retinopathy of Prematurity)
Маргарита Потягайло
Завідуюча офтальмологічним відділенням
Волинського обласного дитячого територіального медичного об’єднання
Ретинопатія недоношених (РН) – тяжке вітреоретинальне захворювання очей (вазопроліферативна ретинопатія), що виникає переважно у глибоко недоношених, незрілих дітей.
Вперше захворювання описане Т. Теrrу (1942) як окрема нозологічна форма, обумовлена недоношеністю, яка отримала назву ретролентальна фіброплазія (РЛФ). Основним її симптомом була наявність сіро-білих мембран, плівок відразу за кришталиком в скловидному тілі, що фактично являє собою далеко розповсюджений процес (V стадія ретинопатії недоношених за сучасною класифікацією).
В 1984 році Раtz з співавторами встановили зв’язок ретролентальної фіброплазії (РЛФ) з застосуванням кисню у новонароджених дітей. Перша епідемія РЛФ, що стала причиною сліпоти в 30% випадків втрати зору у дітей дошкільного віку в кінці 1940-х років, виникла в період відносно безконтрольного застосування оксигенотерапії у недоношених дітей. Проте, не дивлячись на посилення контролю за оксигенотерапією, друга епідемія РЛФ, яку називають тепер ретинопатією недоношених (РН), виникла в кінці 1970-х років і продовжувалась в 1980-і роки.
Завдяки успіхам неонатології у всіх розвинутих країнах значно збільшилось число виживаючих глибоко недоношених і раніше нежиттєздатних дітей. Вважають, що в сучасних умовах більше 50% 25-тижневих плодів з масою тіла 700 г мають можливість вижити. Ретинопатія недоношених (РН) розвивається серед виживших недоношених дітей в 9-46,9%, а серед глибоко недоношених з масою тіла при народженні менше 1000 г – у 69-90%.
Не дивлячись на значні досягнення у виявленні і лікуванні РН, в теперішній час вона стала однією з причин сліпоти і слабозорості з раннього дитинства у розвинутих країнах. Так, в США щорічно близько 500 дітей сліпнуть внаслідок РН, а у Великобританії реєструють 50-100 випадків розвитку сліпоти на рік.
Клінічні прояви та класифікація РН:
В основі клінічних проявів РН лежить порушення васкулогенезу сітківки. В сітківці, що нормально розвивається, судини відсутні приблизно до 16 тижнів гестації. В цей період кисень поступає в сітківку шляхом дифузії з капілярів хоріоідеї, і після 16 тижнів починається розвиток власних судин сітківки, і завершується цей процес лише перед народженнням дитини (40 тиж.).
Практично у всіх недоношених дітей є офтальмоскопічні відмінності від доношених дітей. На очному дні недоношених (в нормі) завжди виявляють аваскулярні зони на периферії сітківки, причому їх розміри тим більші, чим менший гестаційний вік дитини на момент огляду. Наявність таких аваскулярних зон на периферії очного дна – це не прояв РН, а лише свідоцтво недорозвитку сітківки, незавершеності васкулогенезу і, відповідно, можливості розвитку ретинопатії в подальшому.
В своєму розвитку РН проходить декілька стадій, що відображають прогресування активного процесу. На зміну активній РН приходить стадія регресу, а потім — рубцева стадія захворювання.
В 1984 році в Канаді офтальмологами з 11 ведучих країн світу була розроблена Міжнародна класифікація активної ретинопатії новонароджених і єдина форма реєстрації патологічних змін в оці. Цю класифікацію з невеликими уточненнями і доповненнями використовують до теперішнього часу. Згідно класифікації активну РН поділяють в залежності від стадії, локалізації і поширеності процесу.
Стадія 1 — поява демаркаційної лінії на межі судинної і безсудинної сітківки. Білувата лінія, розміщена в площині сітківки, являє собою скупчення гіперплазованих веретеноподібних клітин. Перед демаркаційною лінією судини розширені, звивисті, утворюють аномальні розгалуження, аркади, і, раптово обриваючись, не проникають в безсудинну сітківку периферичніше від лінії.
Стадія 2 – поява валу (чи гребеня) на місці демаркаційної лінії. Сітківка в цій зоні потовщується і промінує в скловидне тіло у вигляді жовтуватого валу. Судини перед валом різко розширені, звивисті, хаотично діляться, утворюють артеріовенозні шунти. Сітківка в цій зоні набрякає. Виявляють також набряк і судинні порушення в перипапілярній ділянці.
При 1 і 2 стадіях у 70-80% хворих РН можливий самовільний регрес захворювання з мінімальними залишковими змінами на очному дні.
Стадія 3 – це поява екстраретинальної фіброваскулярної проліферації в ділянці валу; посилюється судинна активність в задньому полюсі ока і ексудація в скловидне тіло, з’являються пучки новоутворених судин, щільної тканини за межами сітківки позаду валу.
При невеликій розповсюдженості процесу (1-2 годинних меридіани), так як і в перших двох стадіях, можливий самовільний регрес, хоч залишкові явища на периферії сітківки лишаються.
Розвиток екстраретинального процесу на 5 сусідніх або 8 сумарних меридіанах називають пороговою стадією РН, коли процес прогресування стає практично незворотнім і вимагає термінових лікувальних заходів, не пізніше 72 год.
Стадія 4 – часткове відшарування сітківки. Поділяється на IVа (без поширення процесу на макулярну область) і ІVб (з відшаруванням в макулі).
Відшарування і тракції сітківки є наслідком проникнення новоутворених судин в скловидне тіло з фіброзом і рубцюванням.
Стадія 5 – повне або тотальне відшарування сітківки, воронкоподібної форми, з вираженою деструкцією скловидного тіла.
Стадії 4 і 5 РН називають термінальними, в зв’язку з поганим прогнозом і різкою втратою зорових функцій.
Поділ процесу по розповсюдженості і локалізації має практичне значення лише для перших трьох стадій РН. Розповсюдження процесу на очному дні оцінюють за годинниковими меридіанами (від 1 до 12). За локалізацією РН виділяють 3 зони. Зона 1 – коло з центром в диску зорового нерва (ДЗН) і радіусом, що дорівнює подвійній відстані диск-макула; зона 2 – кільце периферичніше першої зони до зубчатої лінії в носовому сегменті; зона 3 – півмісяць на темпоральній периферії, зовні від 2-ї зони.
РН в зоні 1 протікає значно тяжче і має гірший прогноз.
Окремо виділяють прогностичну неблагоприємну форму активної РН, названу як “плюс-хвороба”. Для “плюс-хвороби” характерний ранній початок і швидке прогресування; в процес, як правило, втягується зона 1, тобто задній полюс ока. “Плюс-хвороба” протікає з різкою судинною активністю та різким розширенням судин на диску зорового нерва, крововиливами і вираженою ексудативною реакцією, розвивається ригідність зіниці, неоваскуляризація райдужки, спостерігається ексудативний випіт в скловидне тіло. Якщо ці симптоми продовжують наростати, то до 5-7 діб може виникнути відшарування сітківки.
Тривалість активної РН в середньому 3-6 місяців. Вона завершується або спонтанним самовільним регресом в перших двох стадіях, або фазою рубцювання (3-я стадія) з залишковими змінами в очному дні різного ступеня вираженості. В рубцьовій стадії можуть бути виявлені зміщення магістральних судин, їх звивистість, перерозподіл пігменту, зони атрофії сітківки, формування пре-, суб- і інтраретинальних мембран, розривів сітківки, тракційної деформації диску зорового нерву, ектопії і деформації макули, формуються серповидні складки сітківки, тракційне відшарування сітківки.
Проф. Хватовою О.В. з співавторами в 1998 році запропонована класифікація регресивної і рубцьової стадії РН, яка теж має 5 ступенів, як і активна РН.
- 1 ступінь – мінімальні судинні і інтраретинальні зміни на периферії очного дна, що майже не впливають на зорові функції.
- 2 ступінь – ектопія макули і вітреоретинальні дистрофічні зміни на периферії, що можуть призвести до вторинних відшарувань сітківки.
- 3 ступінь – груба деформація ДЗН з вираженою ектопією і дистрофією макулярної ділянки, що поєднується із змінами на периферії очного дна.
- 4 ступінь – наявність грубих серповидних складок сітківки із значним порушенням зорових функцій.
- 5 ступінь – тотальне воронкоподібне відшарування сітківки. На відміну від 5-ї стадії активної РН, відшарування сітківки при рубцьовій РН завжди має тракційний характер.
Диференціальна діагностика:
Диференціальна діагностика в активних стадіях не викликає значних затруднень. “Плюс-хворобу” слід диференціювати від ретинобластоми; іноді від ретинальних геморагій новонароджених, які частіше бувають при ускладнених пологах у доношених дітей із значною масою тіла.
Рубцьові стадії РН складно диференціювати від: первинного персистуючого гіперпластичного скловидлного тіла (ППСТ); периферичного увеіту; Х-хромосомного ретиношизису; сімейної ексудативної вітреоретинопатії. Остання розвивається в більш старшому віці і не має зв’язку з недоношеністю.
Патогенез:
Патогенез РН, не дивлячись на проведення багаторічних клінічних і експериментальних досліджень, до кінця не вивчений. Сучасні уяви про РН зводяться до визнання мульфакторності її походження, коли безліч різноманітних факторів ризику викликають порушення нормального васкулогенезу сітківки у глибоко недоношених, незрілих малюків.
Недоношена дитина народжується з незавершеною васкуляризацією, особливо периферії сітківки (яка починається з 16 тижнів гестації і завершується при народженні в 40 тижнів). Після передчасного народження дитина попадає з умов внутріутробної гіпоксії у відносну гіпероксію нормального повітря або отримує додатково кисень, який може стати основою для порушення нормального васкулогенезу сітківки. Раніше вважалось, що надмірна кількість кисню негативно впливає на ендотелій незрілих капілярів, викликає їх вазооблітерацію, що призводить до гіпоксії сітківки і наступному аномальному ангіогенезу. Експериментальні дослідження останніх років доказують переважну роль кисню в розвитку РН при зміні фаз гіпероксії/гіпоксії. Згідно до цієї гіпотези гіпероксигенація викликає звуження просвіту капілярів, що при тривалій гіпероксії в кювезах призводить до запустіння і облітерації незрілих судин з наступним розвитком проліферації ендотелію і ростом новоутворених судин.
Одним з механізмів патогенезу РН є вплив вільних радикалів на мембранні структури сітківки і її судини. Сприяють розвитку РН захворювання матері, що викликають гіпоксію плода: хронічні хвороби жіночих статевих органів, гестоз, кровотеча в родах, хронічні інфекції, куріння, прийом бета-блокаторів та інше.
Згідно сучасним даним, оксигенотерапія також є одним із важливих факторів ризику розвитку РН. Аналіз інтенсивності кисневої терапії свідчить, що фактором ризику є перебування дитини в умовах штучної вентиляції легень (ШВЛ) більше 5 днів, тривалості загальної оксигенотерапії більше 20 днів, парціального тиску кисню в крові >80 мм рт. ст.
Встановлена залежність розвитку РН від наявності у недоношених ацидозу, сепсису, анемії, повторних гемотрансфузій та інше. Надлишок вільних радикалів викликають такі стани, як бронхо-легенева дисплазія, респіраторний дистрес-синдром, кардіопатія, які негативно впливають на мембранні структури сітківки та її судини.
Важливим є питання впливу світла на перебіг РН. В природніх умовах васкулогенез сітківки завершується при відсутності світлового впливу, а недоношена дитина потрапляє в умови надлишку світла на незрілу сітківку. Проте переконливих доказів впливу надлишку світла на виникнення і перебіг РН не отримано.
В 1992 році І. Flimn висунув гіпотезу про генетичну зумовленість РН, що розвиток РН зв’язаний з пошкодженням генетичної програми васкулогенезу сітківки, ймовірно, ще в період внутріутробного розвитку, а саме захворювання розвивається після народження дитини. На користь цієї гіпотези свідчать і результати досліджень із застосуванням методів молекулярної генетики.
Відомо, що зчеплена зі статтю сімейна ексудативна вітреоретинопатія фенотипічно схожа з РН і в ряді випадків зв’язана з мутацією гену хвороби Норрі.
Діагностика:
Виявлення ретинопатії недоношених проводиться офтальмоскопією, що виконується досвідченим офтальмологом.
Американська асоціація неонатологів рекомендує оглядати всіх недоношених дітей, гестаційний термін яких менший 30 тижнів і маса тіла <1300 г, незалежно від загального стану і тривалості оксигенотерапії, а дітей до 35 тижнів з масою тіла до 1800 г – лише у випадках проведення лікування киснем.
За даними Хватової О.В. із співавторами (Москва) в групу ризику по РН слід включати всіх недоношених з масою тіла менше 2000 г при строках гестації менше 35 тижнів незалежно від соматичної обтяженості.
Оптимальні строки першого огляду варіюють від 3 до 7-9 тижнів після народження, бо мінімальний гестаційний вік, коли виникають перші прояви РН, складає 30-34 тижні.
При виявленні РН наступні огляди рекомендовано проводити щотижня, а при активному процесі з ознаками “плюс-хвороби” – кожні 2-3 дні. Але і при відсутності симптомів хвороби повторні офтальмоскопії слід проводити кожні 3-4 тижні до того часу, поки не дозріє сітківка.
Важливою є методика проведення профілактичного огляду. Основну увагу слід звертати на детальний огляд периферії очного дна при доброму мідріазі, з застосуванням непрямої бінокулярної офтальмоскопії, а також застосовуючи нетравматичні повороти очного яблука в потрібному напрямку. Зменшенню травматичності огляду сприяє застосування спеціальних повікорозширювачів для недоношених. Можливе застосування дитячих повікопідйомників при збереженні всіх застережень.
Ригідність зіниці недоношених значно затруднює адекватний огляд периферії очного дна, а виникнення побічних системних ускладнень на різні мідріатики часто обмежує їх вибір. Найкращий мідріаз можна досягти 1-2% розчином тропікаміду (мідріацилу) в комбінації з 2,5% розчином фенілепінефрину.
Замість тропікаміду можна застосовувати 0,1% розчин атропіну. Фенілепінефрин можна замінити 0,1% адреналіном чи 1% мезатоном.
Інстиляції проводять за 1 годину до огляду. Можливі побічні реакції: тахікардія, підвищення АТ (частіше від епінефрину і адреналіну).
Діагноз ретинопатії недоношених відображує активність, стадію, зону ураження сітківки, як приклад: активна ретинопатія недоношених, праве око – стадія 2-3, поширеність 8-12, зона 2 (внутрішня); ліве око – стадія 2, поширеність 10, зона 2 (зовнішня).
Лікування:
Лікування РН є надзвичайно складною проблемою. Консервативне лікування в 1-2, 3-а стадії може включати застосування вітаміну Е, інших антиоксидантів, судиноукріплюючих препаратів, емоксипіну, хоча більшість авторів рекомендує ретельне спостереження за дітьми з 1 -2 активною стадією, яка в 90% спонтанно регресує. Згідно існуючим на сьогоднішній день даним при стадії 3(+) спонтанний регрес буває лише у 50% випадків.
Основним є хірургічне лікування РН, яке поділяють на профілактичне і реабілітаційне.
Профілактичне лікування – це кріотерапія і лазеркоагуляція (транссклеральна і транспупілярна), показами до яких є порогова (3-я) стадія активної РН з поширеністю 5 годинникових меридіанів підряд чи 8 – сумарно, а також всі процеси в зоні 1 очного дна або “плюс-хвороба”. Обидві методики вперше були випробувані майже одночасно в Японії. В 1968 році М. Nagata з співавторами повідомили про перші позитивні результати ксенонової фотокоагуляції; кріокоагуляцію вперше застосував О. Yamachita в 1972 р. Терапевтична дія лазерної і кріокоагуляції пояснюється деструктивною дією на ішемічні зони аваскулярної сітківки.
На теперішній час лазерна коагуляція – процедура вибору для проведення профілактичного лікування, її переваги перед кріокоагуляцією полягають в можливості кращого дозування ступеню коагуляції і формування більш ніжних рубців на сітківці, меншій частоті офтальмологічних ускладнень, більших можливостей при лікуванні 1 зони.
Проте виконання процедури є досить важким при ригідній зіниці, вимагає тривалого часу для нанесення коагулятів (250-2500) на протязі 15-45 хвилин, в умовах загального знеболення.
Ефективність лазерного лікування при РН – 73-90%, кріокоагуляції – 50-79% за даними різних авторів. В 2002 році в м. Києві на базі Київського центру мікрохірургії ока створено Республіканський центр по лазерному лікуванню ретинопатії недоношених.
При неадекватності профілактичного лікування та розвитку тяжких рубцьових стадій РН показана реабілітаційна хірургія: ленсвітректомія, ленсзберігаюча хірургія, різні методики склерального вдавлення при відшаруваннях сітківки, з додатковим висіченням мембран і введенням силікону чи саморозширюючих газів в скловидне тіло.
Функціональні результати хірургічних втручань залишають бажати кращого. Після ленсвітректомії гострота зору рідко перевищує 0,01; в більшості випадків лише покращується характер світловідчуття, з’являється можливість слідкування за предметами біля обличчя і орієнтації в приміщенні.
Література:
- Клініка Віллса: діагностика і лікування очних хвороб /За ред. Д. Каллома та Б. Чанга.- Пер. з англ.- Львів: Медицина світу, 1999.- 492 с.
- Матеріали II конференції дитячих офтальмологів України.- Судак, 2003.
- Матеріали науково-практичної конференції “Актуальні питання дитячої офтальмології”.- Москва, 1997.
- Теймор Д., Хойт К. Детская офтальмология.- ЗАО “Издательство БИНОМ”, 2002.
- Федоров А. А. Пренатальное развитие сосудов сетчатой оболочки глаза человека //Вестник офтальмологии.- 2003.- N 4.- С.59-63.
- Шамшинова А.М. Наследственные и врожденные заболевания сетчатки и зрительного нерва.- Москва, 2001.
Переглянуто редакційною колегією I.B.I.S.: 13/10/2004
Статевий розвиток дівчат (продовження)
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”
Статевий розвиток – ріст грудей в жінок

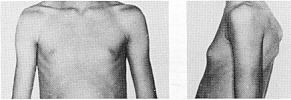

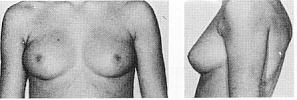
дорослому типу, контур ареоли переходить
в загальний контур груді.
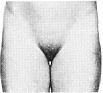
Статевий розвиток – ріст лобкового волосся
Стадія 1. Відсутність лобкового волосся.
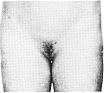
Стадія 3. Волосся стає помітно темнішим, жосткішим та більш курчавим. Волосся повільно поширюється по лобку.
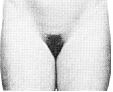

Ілюстрації подані за згодою J.M. Tanner. Tanner JM: Growth at Adolescence, ed. 2, Blackwell Scientific Publications, 1982.
Синдром нігтів-надколінника
(Nail Patella Syndrome)
І.М. Дубчак
Лікар-генетик
Хмельницької медико-генетичної консультації
Синоніми:
- Оніхоостеодисплазія;
- Хвороба Фонга (Fong disease);
- Хвороба Іліака-Горна (Iliac Horn disease);
- Синдром Тернера-Кізера (Turner-Kieser).
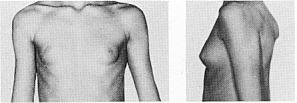
Основні діагностичні критерії:
Гіпоплазія нігтів, надколінника, захворювання нирок.
Клінічна картина:
Нігті вражаються у 80-90% випадків. Найчастіше вражаються І, ІІ та ІІІ пальці. Нігті вузькі, гіпопластичні, подовжені трикутні ложа, знебарвлені, розшаровуються (мал. 1, мал. 2). Надколінник уражається у 60% випадків. Він є гіпоплазований, може мати форму багатокутника або взагалі бути відсутнім. Відмічається вивих надколінника. Всі ці зміни асоціюються із остеоартритом, остеоартрозом, кровотечею в порожнину колінного суглоба. Обмежена рухливість в ліктьових суглобах, що може бути пов’язане із ліктьовим птеригіумом.
Рентгенологічно: Голівка променевої кістки недорозвинута і зміщена дозаду. Спостерігаються нарости на задній поверхні здухвинної кістки, симетричні, двобічні, від маленьких до дуже великих розмірів. Інколи буває сколіоз. Захворювання ускладнюється варусною деформацією гомілки.
Найчастіше спостерігається нефропатія, гематурія, протеїнурія, що супроводжується набряками, нирковою гіпертензією. Рідше буває сечокам’яна хвороба, вроджені вади нирок – везікоуретральний рефлюкс, агенезія, гіпоплазія, подвоєння. Порушення функції нирок супроводжується такими ускладненнями: нефрит (набряки, гіпертензія, азотемія, олігурія), нефроз (набряки, гіпоальбумінемія, гіперліпідемія), хронічна ниркова недостатність.
Менш розповсюдженими є такі ознаки цього захворювання як дефекти верхньої губи та піднебіння, незвичайні аномалії скелету. Такі аномалії включають погано розвинуті лопатки, клинодактилію, клишоногість, сколіоз та вади кісток хребта.
У пацієнтів також часто зустрічається катаракта, птоз, або корнеальні проблеми, такі як глаукома.
Обстеження:
Проводиться рентгенологічне обстеження рук, ніг, хребта, тазу. Можна виявити зміни в променевій кістці ліктьового суглобу, відростки на здухвинних кістках, гіпоплазію надколінника, сколіоз. Ниркова патологія підтверджується аналізами сечі, визначають ескрецію креатиніну, протеїнурію, протеїново-креатиновий індекс (в нормі він менший ніж 0,2). Проводять біохімічне дослідження крові з визначенням рівня сечовини, креатиніну.
Важливим дослідженням є біопсія та УЗД нирок. Клубочки нирок змінені, клітинні мембрани потовщені до повного або часткового гломерулосклерозу. Сонографія допомагає краще оцінити неоссифікований надколінник у маленьких дітей.
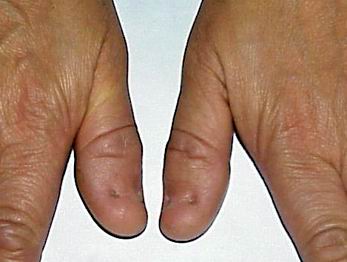
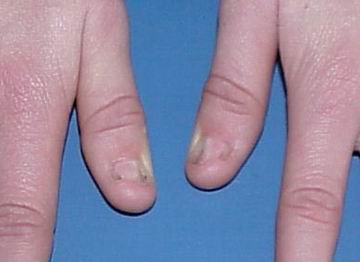
Пренатальна діагностика:
У літературі існують поодинокі описи пренатальної ультразвукової діагностики синдрому нігтів-надколінника у третьому триместрі. Однак загалом пренатальна діагностика неінформативна.
Диференційний діагноз:
Проводиться із синдромом маленького надколінника, синдромом аплазії-гіпоплазії надколінника, синдромом Тернера.
Етіологія:
Мутація в гені LMX1B, який кодує протеїн, що є дуже важливим у організації ембріонального розвитку кінцівок, і локалізований на довгому плечі 9 хромосоми 9q34.
Частота виникнення:
1:50000 новонароджених. Поширене у всьому світі та у всіх етнічних групах. Найбільший ризик виникнення цього синдрому мають ті особи, у сім’ях яких вже зустрічалось це захворювання.
Співвідношення статей: Ч1 : Ж1.
Тип успадкування:
Аутосомно-домінантний з повною пенетрантністю та різною експресивністю.
Патогенез:
Порушення функції нирок є результатом вторинної гломерулопатії. У нирках плоду і дорослих людей виявляється високий рівень експресії гену LMX1B, тому припускають, що він має значний вплив на розвиток нирок. Втрата функції одного алеллю цього гену є основною причиною випадків нефропатії. Патологія кісткової системи обумовлена розпадом колагену, а також порушенням синтезу, всмоктування і перетворення колагену.
Історія хвороби:
Захворювання проявляється в дитинстві. Поступово прогресують ознаки порушень функції нирок. Змінюється хода, постава, зменшується рухливість суглобів, шкіра стає бліда, еластичність її зменшується. Для хворих характерний низький зріст, худорлявість, порушення фізичного розвитку. Дослідження показують, що пацієнти з синдромом нігтів-надколінника мають ризик розвитку глаукоми, яка може бути діагностована в будь-якому віці. Для таких людей необхідні щорічні огляди окуліста з контролем гостроти зору, очного дна та тиску.
Синдром нігтів-надколінника також асоціюється з аномаліями рогівки, катарактами та астигматизмом.
Прогноз:
В цілому сприятливий, але у 100% випадків розвивається хронічна ниркова недостатність. 30-35% хворих помирають від ниркової недостатності. Протеїнурія – перший симптом прогресування хвороби.
Лікування:
Проводиться консервативне і хірургічне лікування. Використовують препарати кальцію та вітамін D для регуляції рівня кальцію в крові. При набряках призначають діуретики та препарати, що регулюють водно-сольовий баланс, іноді – кортикостероїдні гормони. Хірургічне лікування полягає в пластиці суглобів (надколінника), відсмоктуванні рідини з порожнини суглобів. Антибактеріальна терапія проводиться у випадках загострення артритів, гломерулонефритів. При прогресуванні хронічної ниркової недостатності хворі переводяться на апарат “штучна нирка” та часом потребують трансплантації нирок. Деяким пацієнтам необхідне хірургічне лікування катаракти, глаукоми.
Профілактика:
Методом запобігання синдрому нігтів-надколінника є генетичне консультування з ретельним вивченням родоводу, робота з подружжям, у родоводі якого виявлений цей синдром.
Пренатальна діагностика мало інформативна. Дуже важливим моментом є профілактика ускладнень, що досягається своєчасним лікуванням проявів хвороби. Хворі знаходяться під спостереженням педіатрів, терапевтів, нефрологів, ортопедів-травматологів, окулістів.
Номер з каталогу МІМ:
161200 Nail-Patella Syndrome; NPS.
Література:
- Chen H, Lun Y, Ovchinnikov D, Kokubo H, Oberg KC,Pepicelli CV, Gan L, Lee B, Johnson RL. Limb and kidney defects in Lmx1b mutant mice suggest an involvement of LMX1B in human nail patella syndrome. Nature Genet 1998;19:51-55.
- Daniel CR, Osment LS, Noojin RO. Triangular lunulae: a clue to nail-patella syndrome. Arch Derm 1980;116:448-449.
- Hawkins CF, Smith OE. Renal dysplasia in a family with multiple hereditary abnormalities including iliac horns. Lancet 1950;I:803-808.
- Leahy MS. The hereditary nephropathy of osteo-onychodysplasia (nail patella syndrome). Am J Dis Child 1966;112:237-241.
- Looij BJ, Te Slaa RL, Hogewind BL, van de Kamp JJP. Genetic counselling in hereditary osteo-onychodysplasia (HOOD, nail-patella syndrome) with nephropathy. J Med Genet 1988;25:682-686.
- Mark TM, Rywlin AM, Unger H. Cystic adventitial degeneration of the popliteal artery: its occurrence in a patient with the nail-patella syndrome. Arch Path Lab Med 1983;107:186-188.
- Morita T, Laughlin O, Kawano K, Kimmelstiel P. Nail-patella syndrome. Arch Intern Med 1973;131:271-277.
- Sabnis SG, Antonovych TT, Argy WP, Rakowski TA, Gandy DR, Salcedo JR. Nail-patella syndrome. Clin Nephrol 1980;14:148-153.
Переглянуто редакційною колегією I.B.I.S.: 31/05/2005
Синдром Нагера
(Nager syndrome)
Т.Д. Загорулько
Завідуюча відділенням недоношених новонароджених
Волинського обласного дитячого територіального медичного об’єднання
Синдром Нагера (акрофаціальний дизостоз) – рідкісне генетичне захворювання, яке включає в себе фізичні аномалії. Вперше синдром описаний в 1948 р. F. Nager і J. de Reynier.
Діагностичні критерії:
Гіпоплазія нижньої щелепи, антимонголоїдний розріз очей, стеноз або атрезія слухового проходу, гіпо- або аплазія променевої кістки і І пальця кисті.
Частота:
Популяційна частота невідома, за даними літератури описано більше 90 випадків.
Етіологія:
Більшість випадків – спорадичні, однак є описання випадків, коли у хворих батьків народжувались хворі діти, що може свідчити про аутосомно-домінантний тип успадкування; а також народження хворих дітей у здорових батьків, що не виключає аутосомно-рецесивний тип успадкування.
Клініка:
Психомоторний розвиток дітей звичайно сповільнений. Кондуктивна глухота двобічна, що зумовлює проблеми з артикуляцією.
Краніофаціальні аномалії включають: антимонголоїдний розріз очей, високе перенісся, мікрогнатію, часткову або повну відсутність нижніх вій, преаурікулярні вирости, низько розміщені вушні раковини, атрезію зовнішнього слухового каналу, розщілину піднебіння, губи.
Кінцівки: гіпоплазія або аплазія І пальця кисті, з наявністю чи відсутністю променевої кістки, проксимальний променеволіктьовий синостоз, обмеження розгинальних рухів в лікті, коротке передпліччя.
Поєднані аномалії:
Розумова відсталість, дефіцит росту, колобома нижньої повіки, протяжні зони росту волосся голови до щік, розщелина губи, гіпоплазія гортані чи надгортанника, фіброз і анкілоз скронево-щелепного суглобу, синдактилія, клинодактилія, камптодактилія, подвійний і трьохфаланговий І палець кисті, відсутність або гіпоплазія пальців ступні, синдактилія та находження пальців ступні, вальгусний та широкий великий палець ступні, вкорочення кінцівок, клишоногість, гіпоплазія І ребра, сколіоз, аномалії шийного відділу хребта, вади серця (тетрада Фалло, ДМШП, відкрите овальне вікно), мікроцефалія, гідроцефалія, стеноз лікворних шляхів, полімікрогірія, сечостатеві аномалії, хвороба Гіршпрунга, гастрошизис, уртикарна висипка, неправильне прорізування зубів, каріозні зуби.
Пренатальна діагностика:
Важкий ступінь синдрому Нагера можна діагностувати за допомогою ультразвукового дослідження в 16 тижнів вагітності. Рекомендована генетична консультація.
Диференціальний діагноз:
Нижньощелепно-лицевий дизостоз, окуло-аурікуло-вертебральна дисплазія, синдром Халлермана-Штрайфа, можливе поєднання синдрому Нагера з 45,Х моносомією.
Прогноз:
Залежить від ступеню важкості даного захворювання. При тяжкій формі (виражені лицеві аномалії, вкорочені кінцівки, відсутні пальці) – більшість дітей не життєздатні. Перинатальна смертність приблизно 20% і пов’язана з вторинними дихальними розладами. Спотерігається високий ризик невиношування вагітності. При середній формі пораження обличчя і вух є подібним до Treacher Collins синдрому. У деяких дітей можуть виникати труднощі з прийманням їжі, диханням. Відхилення в мовному розвитку пов’язані з первинною втратою слуху.
Акушерська тактика:
При діагностуванні даної патології пренатально і наявності поєднаних важких вад розвитку у плода, необхідна консультація генетика з подальшим виробленням тактики про доцільність виношування чи переривання даної вагітності. Відразу після народження необхідно забезпечити дитині адекватне дихання.
Тип успадкування:
Ймовірно аутосомно-домінантний.
Лікування:
Після народження негайна увага повинна бути звернена на функцію дихання та приймання їжі, інколи виникає потреба в накладанні трахеостоми та гастростоми; годування через стому, а також зондове годування можуть проводитись тривалий час, якщо рухи щелепи сильно обмежені. Пізніше, але якомога раніше слід починати оральну дачу їжі, корегувати функцію слуху, проводити мовну стимуляцію, підключати артикуляцію. Фтористе лікування і обмеження кількості цукру в їжі є необхідним для зменшення пошкодження зубів. У віці 3-4 років необхідно зменшити нижньощелепний анкілоз шляхом накладання шин та за допомогою функціональної терапії. Пружинне витягування може мати успіх. В 6-7-річному віці, або пізніше слід починати подовження віток нижньої щелепи – це може бути здійснено шляхом трансплантації кістки, або витягування. Лікування повинно здійснюватись після прорізування постійних зубів. Спочатку реконструктивні втручання здійснюються на нижній щелепі, потім верхній та скуловій кістці. Сколіози корегуються за допомогою фізичних вправ. Гнусавість мови залишається однією з проблем. Не завжди вдається скоригувати її хірургічним шляхом через відсутність м’якого піднебіння; накладання обтуратора не завжди можливе по причині неправильного розміщення зубів.
Номер з каталогу МІМ:
154400 Acrofacial Dysostosis 1, Nager Type; AFD1.
Література:
- Козлова С.И., Демикова Н.С., Семанова Е., Блинникова О.Е. Наследственные синдромы и медико-генетическое консультирование.- М.: Практика, 1996.- С.20.
- Acrofacial Dysostosis 1, Nager Type; AFD1. OMIM #154400.
- Gorlin RJ., Cohen MM., Hennekam RCM. Syndromes of the Head and Neck.- 4th Edition. Oxford University Press. 2001:802-805.
- Jones KL. Smith’s Recognizable Patterns of Human Malformation.- 5th Edition. W.B. Saunders Company. 1997:258-259.
- Vargervik K. Mandibular malformations: growth characteristics and management in hemifacial microsomia and Nager syndrome. Acta Odontol Scand 1998 Dec;56(6):331-8.
Переглянуто редакційною колегією I.B.I.S.: 19/10/2002
|
|
|
|
|
|





