
Serhiy
Рото-лицьові розщілини
(Orofacial Clefts)
О. О. Михасюк
Спеціаліст з інформаційного забезпечення
Рівненського ОМНІ-Центру
Рото-лицьові розщілини – це отвори на обличчі в області рота. Ці отвори можуть утворюватися на губах, на твердому піднебінні, або на тканинах задньої частини рота (м’якому піднебінні). Існує два основних типи рото – лицьових розщілин. Перший – це розщілина губи, яка зазвичай вражає і частину твердого піднебіння. Така вада називається “розщілина губи та піднебіння”. Другий тип – це так звана “ізольована розщілина піднебіння”, що виникає лише на твердому піднебінні, не вражаючи при цьому губ.
У викладеній нижче інформації ми використовуємо термін “рото-лицьова розщілина” для позначення обох типів вад. Існує понад 400 синдромів, при яких в дитини можуть виникати рото-лицьові розщілини поряд із проявами інших вроджених вад. Ми не мали за мету висвітлення цих проблем в даному інформаційному листку.
Коли і як часто виникають рото-лицьові розщілини?
Зазвичай, такі аномальні отвори на обличчі утворюються на ранньому етапі розвитку плоду. Губи змикаються на 5-6 тижні після зачаття, тверде піднебіння на 10 тижні. Частота виникнення незрощення губи/піднебіння становить приблизно 1 на 1000 новонароджених (дані по європейській расі). Серед представників азіатської раси цей показник становить 1,7 на 1000 новонароджених. Найвищим він є серед американських індіанців (3,6 : 1000), найнижчим серед афро-американців (1 на 2500 новонароджених). До того ж, в чорношкірих новонароджених хлопчиків ця вада виникає частіше, ніж у дівчаток.
Ізольована вада твердого піднебіння виникає менш часто (1 : 2000 новонароджених). На відміну від розщілини губи/піднебіння, ізольовані вади твердого піднебіння виникають з приблизно однаковою частотою серед представників різних рас. Інша відмінність полягає у тому, що подібні вади частіше вражають саме представників жіночої статі.
Що спричиняє розщілини губи/піднебіння?
Причини виникнення розщілини губи/піднебіння ще до кінця не з’ясовані. На виникнення цих вад можуть впливати різноманітні фактори: генні порушення, вплив зовнішнього середовища, вживання певних медикаментів (протисудомних препаратів), інфекції, хронічні хвороби матері, вживання матір’ю алкоголю під час вагітності, дефіцит вітаміну В (фолієвої кислоти).
Як вже зазначалося, розщілини губи/піднебіння можуть виникати як самі по собі, так і у супроводі інших вроджених вад розвитку – явних або прихованих. Приблизно 13% дітей із розщілинами губи/піднебіння мають інші ВВР. Є випадки, коли розщілини губи/піднебіння супроводжуються певними генетичними синдромами, які створюють додаткові ускладнення під час лікування уражених дітей та несуть у собі потенційну загрозу повторного виникнення у інших членів родини. Отож, діти із розщілинами губи/піднебіння мають бути уважно оглянуті лікарем одразу після народження.
Чим відрізняються причини виникнення ізольованих розщілин піднебіння?
Причини виникнення ізольованих розщілин піднебіння також невідомі. Тут можуть відігравати роль як генетичні фактори, так і чинники навколишнього середовища. Під час досліджень не було встановлено зв’язок між вживанням протисудомних препаратів або алкоголем і розщілинами твердого піднебіння. Проте є підстави вважати, що їх може спричиняти дефіцит вітаміну групи В – фолієвої кислоти.
Ізольовані розщілини твердого піднебіння частіше асоціюються із генетичними синдромами. В дітей із цією вадою набагато частіше зустрічаються й інші вроджені вади розвитку. Показник поширеності ускладнень іншими ВВР становить 50%.
Чи є родинна схильність до виникнення рото-лицьових розщілин?
Якщо в батьків, які не мають ніяких проявів можливих аномалій розвитку губ чи твердого піднебіння, народжується дитина із розщілиною і можна визначити причину її виникнення (наприклад, генетичний синдром), то вірогідність мати наступну дитину із цією самою вадою складає від 2 до 4%. Процент ризику є однаковим як для розщілин губи/піднебіння, так і для ізольованих розщілин твердого піднебіння. Слід також зауважити, що цей ризик стосується тільки можливості виникнення такої самої вади. Якщо ж будь-який тип аномалій розвитку губ чи твердого піднебіння є наявним в когось з батьків, які, проте, не мають уражених дітей, ризик виникнення розщілин становить 3-4% на кожну вагітність. Якщо ж ураженими є хтось з батьків, та попередня дитина, то ризик повторного виникнення є значно вищим.
Яким чином розщілини губи/піднебіння уражають дитину?
Розщілини губи можуть відрізнятися за ступенем важкості: це може бути звичайна ямка на губі, а може бути повний отвір, що уражає верхню губу та корінь ніздрів. Також можуть бути ураженими верхні ясна. Розщілини губи можуть виникати по одній та по обидві сторони, вражаючи або не вражаючи тверде піднебіння. Більше 70% дітей із розщілинами губи мають і розщілини піднебіння.
Розщілина піднебіння (поодинока, чи як частина розщілини губи/піднебіння) може вражати як м’яке піднебіння, так і сягати твердого піднебіння. Враженими можуть бути як одна, так і обидві сторони піднебіння.
Чи можна виправити рото-лицьову розщілину?
Для виправлення розщілин губи/піднебіння та ізольованих вад піднебіння роблять спеціальні операції. Час проведення та тип операції залежать від багатьох факторів, а саме: вибору лікаря-хірурга, загального стану дитини та характеру самої розщілини. Більшість хірургів вважає, що кращий вік для проведення операції по виправленню розщілини губи – це тримісячний вік. Розщілини твердого піднебіння також лікуються якомога раніше. Оптимальним віком для цього вважається 9-18 місяців. Часом виникає потреба в проведенні додаткових корегуючих операцій коли дитина досягає більш дорослого віку.
Які специфічні проблеми асоціюються із рото-лицьовими розщілинами?
У дітей із розщілинами часто виникають проблеми із годуванням, хвороби вух та затримка мовленнєвого розвитку, проблеми із зубами. Ступінь вираженості цих проявів може змінюватися і залежить від самої вади та її лікування. Кожна дитина із таким діагнозом потребує індивідуального плану лікування.
Діти із рото-лицьовими розщілинами зазвичай лікуються цілою групою спеціалістів для того, щоб охопити усі аспекти цієї патології. В такі групи входять педіатри, пластичні хірурги, стоматологи, отоларингологи, спеціалісти з проблем затримки мовлення, генетики, психологи та соціальні працівники.
Годування.
Малюки із розщілинами губи не створюють великих проблем під час годування. Вони частіше виникають у дітей із розщілинами губи та піднебіння або ізольованими вадами піднебіння. Саме розщілина на верхній частині рота не дозволяє малюкові нормально смоктати і витягувати молоко з грудних залоз.
Грудне годування таких дітей можливе, але вимагає терпіння та іншого підходу. Воно буває вдалим в дітей із менш важкими проявами.
Більшість лікарських груп скеровують свою увагу саме на розв’язання проблеми годування.
Проблеми із вухами.
Діти із розщілинами твердого піднебіння мають надзвичайну схильність до хвороб середнього вуха. Розщілина може спровокувати утворення у вусі рідини, що може викликати часткову втрату слуху. Якщо рідина містить у собі інфекцію, то в дитини може розпочатися лихоманка та вушні болі. Якщо провести відповідні курси лікування у дитячому та підлітковому віці, втраті слуху можна запобігти.
Протягом перших 3-6 місяців життя усі діти із розщілинами мають бути оглянуті отоларингологом. В разі виявлення рідини в середньому вусі, її можна позбутися або шляхом медикаментозного лікування, або через нескладну операцію. Ця операція полягає в тому, що через вушний канал вводиться тоненька трубка, через яку відсмоктується рідина.
Як рото-лицьова розщілина впливає на розвиток мовлення?
В дітей із розщілинами губи мова є нормальною або майже нормальною. Дехто з пацієнтів із розщілиною твердого піднебіння може навчитися говорити трохи пізніше за своїх однолітків. Вони можуть говорити “в ніс” і нечітко вимовляти деякі звуки. Проте, після проведення операції більшість дітей швидко наздоганяють своїх однолітків і надалі розмовляють нормально.
В чому полягають проблеми із зубами?
В дітей із розщілиною губи та піднебіння виникають особливі проблеми із зубами. Молочні та кореневі зуби можуть бути відсутніми або мати неправильну форму. Для успішного лікування таких випадків необхідна скоординована робота дитячих стоматологів (повсякденне лікування), ортопедів (протезування зубів), лицьових хірургів (оперативна корекція щелеп).
Чи можна запобігти утворенню рото-лицьових розщілин?
Вчені ще мало знають про шляхи запобігання подібних вроджених вад розвитку. В 1995 році було видано результати дослідження, що вказувало на зв’язок між вживанням мультивітамінів із фолієвою кислотою та частотою виникнення рото-лицьових розщілин. Згідно цього дослідження, жінки, які приймали вітаміни мали на 50% менший ризик народити дитину із розщілиною губи та на 25% – із ізольованою вадою піднебіння.
Інші дослідження вказують на наявність генної схильності до виникнення таких вад. Також виникнення розщілин може бути пов’язаним із курінням матері під час вагітності.
Жінкам групи ризику треба уникати вживання алкоголю під час вагітності, що також може спричинити розумову неповноцінність дитини. Ризик виникнення аномалій губ та піднебіння підвищують певні медикаменти (наприклад, від епілепсії). Жінки із хронічними захворюваннями мають проконсультуватися у свого лікаря у період планування вагітності. Усім вагітним жінкам слід вживати лише ті ліки, які їм прописав лікар. Родинам, що відносяться до групи ризику варто звернутися за консультацією до генетика.
Переглянуто редакційною колегією I.B.I.S.: 05/02/2002
Дивіться також:
- Розщілина губи з/без розщілини твердого піднебіння (інформація для спеціалістів)
- Роль фолієвої кислоти у запобіганні розщілини губи і/або піднебіння (інформація для батьків)
Синдром Нунан – зріст дівчат
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”
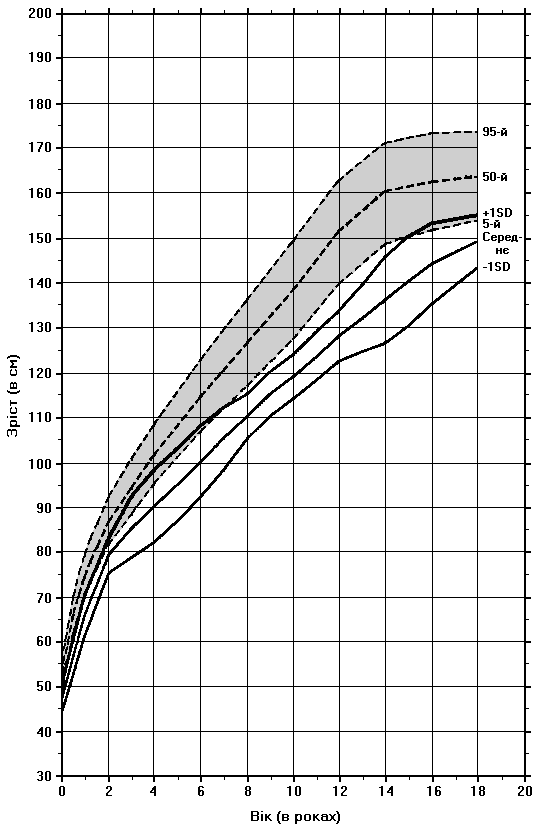
Криві росту дівчат з синдромом Нунан порівнюються з кривими нормального розвитку (пунктирні лінії). Дані отримані в результаті вивчення історій хвороб 48 дівчат з синдромом Нунан. Witt DR et al: Clin Genet 30:150, 1986.
Синдром Рефсума
(Refsum Syndrome)
Д.Р. Ахмеджанова
Інформаційний спеціаліст
Хмельницького ОМНІ-Центру
Вперше дане захворювання було описане норвезьким невропатологом С.Б. Рефсумом у 1945 році.
Що таке синдром Рефсума?
Синдром Рефсума відноситься до генетичних захворювань, що мають назву “лейкодистрофії”, які вражають білу речовину мозку та впливають на моторні функції. Через генетичні порушення у людей із синдромом Рефсума бракує ферменту, який розщеплює фітанову кислоту (це речовина, що зазвичай міститься у їжі, зокрема у молочних продуктах, яловичині, баранині та деяких морських продуктах). Як наслідок, відбувається токсичне накопичення фітанової кислоти у мозку, крові та інших тканинах.
Також існує дитяча (інфантильна) форма синдрому Рефсума, яка спричиняє численні біохімічні порушення, включаючи підвищення рівня концентрації фітанової та прістанової кислот у плазмі. Хвороба з’являється вже на першому році життя. Її проявами є затримка розвитку, порушення слуху та зору, зміни шкіри та специфічні риси обличчя.
Як часто виникає це захворювання?
Синдром Рефсума є рідкісним захворюванням. На даний момент у світовій періодичнй літературі описано близько 100 випадків.
Чи залежить виникнення хвороби від віку, раси та статі?
Даних про залежність від раси та статі немає. Класичний синдром Рефсума проявляється у віці 2-7 років, однак діагноз зазвичай ставиться у дорослому віці. Ознаки інфантильної форми з’являються у ранньому дитинстві.
Які симптоми цього захворювання?
Хвороба зазвичай починається у пізньому дитинстві і супроводжується прогресуючим погіршенням зору, особливо у сутінках (так званою “курячою сліпотою”), яке спричинене дегенерацією сітківки, а також втратою відчуття запаху (аносмія). Інші симптоми, які можуть виникнути в результаті прогресуючої хвороби – це глухота, проблеми із рівновагою та координацією (атаксія), слабкість чи оніміння (периферична невропатія), сухість та лущення шкіри (іхтіоз), порушення серцевого ритму (аритмія). У деяких хворих спостерігаються короткі кістки пальців рук та ніг, або значно коротший четвертий палець ноги. Часом перші ознаки хвороби з’являються тільки у 40-50 років.
Як успадковується це захворювання?
Дане захворювання успадковується за аутосомно-рецесивним типом, тобто для виникнення захворювання необхідна мутація двох копій гену. Кожен з батьків дитини із синдромом Рефсума є носієм однієї копії мутованого гена і кожна їх дитина має 25% ризик успадкування обох генів. Носії не є хворими і не мають жодної ознаки або симптому хвороби.
Який перебіг цього захворювання?
Звичайно перебіг хронічний, з чергуванням періодів ремісії та загострення хвороби. Проте, існують й випадки з відносно швидким летальним кінцем.
До яких терапевтичних (лікувальних) заходів слід вдатись?
Єдиним терапевтично корисним заходом є дієтотерапія. Ідеться про харчовий режим, бідний фітановою кислотою або продуктами, де вона міститься. Виключення фітанової кислоти зовнішнього походження приводить до зниження її рівня у плазмі та тканинах. Тому слід виключити вживання риби, яловичини, баранини, молочних продуктів. Середня кількість щоденного прийому фітанової кислоти – 50-100 мг, в ідеалі її потрібно знизити до 10-20 мг на добу. Слід також обмежити вільне вживання м’яса птахів, свинини, фруктів, овочів.
З якими фахівцями варто проконсультуватись щодо встановлення діагнозу та подальшого нагляду?
Оскільки спостерігаються багато різних симптомів, необхідні консультації таких спеціалістів:
- невролога;
- офтальмолога;
- дерматолога;
- кардіолога.
Більш детальніше про перелік обстежень, які необхідно зробити, Вам розповість Ваш лікар.
Який прогноз можна зробити?
З усіх лейкодистрофій синдром Рефсума найкраще піддається лікуванню, оскільки фітанова кислота не виробляється в організмі, а надходить лише з їжею. Якщо дотримуватися лікування та дієти, то м’язова слабкість, оніміння, сухість та лущення шкіри, зазвичай, зникають. Однак, проблеми із зором та слухом можуть і далі прогресувати, здатність відчувати запахи може не повернутись. Якщо не лікуватись, то синдром Рефсума може призвести до смерті внаслідок порушення серцевої діяльності.
У пацієнтів, які не лікуються, або яким пізно було поставлено діагноз, розвиваються серйозні неврологічні порушення, з якими і пов’язаний високий рівень смертності. Для послаблення проявів неврологічних, офтальмологічних та серцевих порушень необхідно дотримуватись дієти та вдаватись до відповідних терапевтичних заходів.
Переглянуто редакційною колегією I.B.I.S.: 07/07/2005
Синдром Нунан – зріст хлопчиків
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”
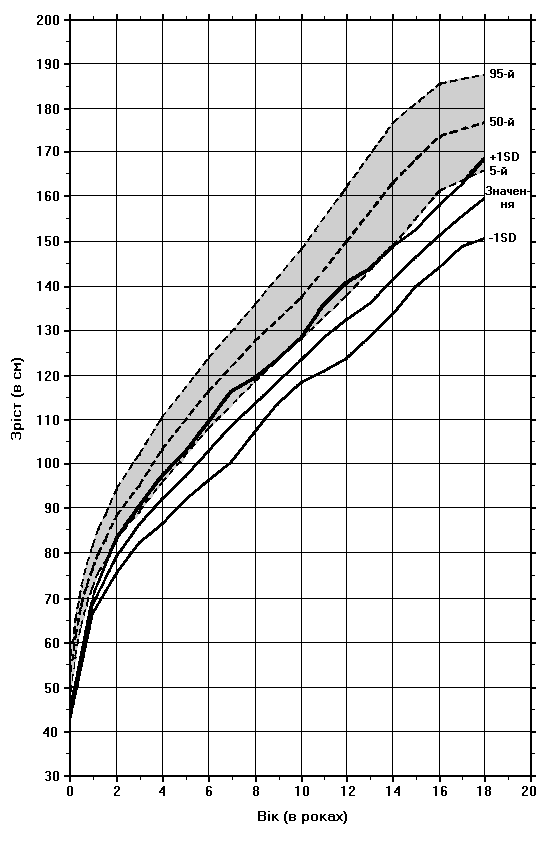
Криві росту хлопчиків з синдромом Нунан порівнюються з кривими нормального розвитку (пунктирні лінії). Дані отримані в результаті вивчення історій хвороб 64 хлопчиків з синдромом Нунан. Witt DR et al: Clin Genet 30:150, 1986.
Синдром Ретта у дітей
(Rett Syndrome)
І.А. Голінко
Лікар-невролог
Дубнівської центральної районної лікарні Рівненської області
Визначення:
Синдром Ретта – одне з найбільш поширених захворювань серед спадкових форм розумової відсталості у дівчаток, назване по імені австрійського педіатра Андреаса Ретта, який вперше його описав у 1966 році.
Основні діагностичні критерії:
Груба затримка психічного розвитку; втрата мови; втрата набутих цілеспрямованих рухів рук; стереотипні рухи рук, які нагадують “миття рук”, стискання, хлопки; порушення ходи (атаксія); затримка темпів росту голови.
Клінічна картина:
В анте- і перинатальному періодах, а також в першому півріччі життя розвиток дітей часто відповідає віку. Однак, в багатьох випадках спостерігається м’язова гіпотонія, незначне відставання в становленні основних рухових навичок (сидіння, повзання, хода), які або не розпізнаються, або недооцінюються.
Вік, в якому вперше відмічаються відхилення у розвитку дітей, – від 4-х місяців до 2,5 років, а найчастіше від 6-ти місяців до 1,5 року. Перші прояви хвороби включають затримку психомоторного розвитку дитини і темпів росту голови, втрату інтересу до гри, дифузну м’язову гіпотонію. Це перша стадія захворювання – стагнація.
Далі наступає період регресу нервово-психічного розвитку, який починається у віці 1-3 років і супроводжується приступами неспокою, крику, порушеннями сну. Впродовж декількох тижнів – місяців дитина втрачає набуті навички, а саме: зникають спрямовані рухи рук, перестає говорити. Одночасно з’являються характерні стереотипні рухи, які нагадують “миття рук”. Можливі судомні приступи, апное, які чергуються з періодами гіпервентиляції. Важливим симптомом є втрата контакту з оточуючими. Ця стадія розвивається так швидко і драматично, що нерідко дітям виставляють діагноз енцефаліту.
Третя стадія охоплює дошкільний і ранній шкільний вік. В цей час стан дітей відносно стабільний. Виражена глибока розумова відсталість, судомні приступи, а також екстрапірамідні розлади: дистонія м’язів, атаксія, гіперкінези. В цей же період приступи неспокою проходять, покращується сон, стає можливим емоційний контакт з дитиною.
В кінці першого десятиріччя життя починається четверта стадія – прогресування рухових розладів. Хворі нерухомі, наростає спастичність, м’язові атрофії і вторинні ортопедичні деформації (сколіоз), з’являються вазомоторні розлади, особливо на нижніх кінцівках, відставання у рості, а у деяких хворих розвивається кахексія. Судоми рідкі, можливе емоційне спілкування з хворим. В такому стані хворі можуть перебувати десятки років.
Основні клінічні ознаки:
Рухи рук. Основні цілеспрямовані рухи рук, такі як маніпулювання іграшками, тримання пляшечки втрачаються в основному до 6-8 місяців, але іноді зберігаються до 3-4 років. Одночасно з’являються стереотипні рухи рук, які спостерігаються весь час, зникаючи під час сну. Ці рухи нагадують “миття рук”, їх стискання, хлопки на рівні грудей, обличчя, може бути смоктання або кусання рук, постукування по грудях або обличчі.
Пізнавальна діяльність. Виражене відставання в інтелектуальному і мовному розвитку.
Атаксія і апраксія. Порушення координації і забруднення в плануванні дій захоплюють як тулуб, так і кінцівки. Спостерігаються порушення рівноваги, тремор, хода на широко розставлених ногах з похитуванням. Деякі хворі не встигають при ранніх проявах хвороби набути навик ходи. Більшість дітей з синдромом Ретта, які вміють ходити, втрачають цю здатність.
Дихальні розлади. Найчастіше зустрічається нерегулярне дихання, приступи гіпервентиляції, апное тривалістю 1-2 хвилини, що достатньо для того, щоб викликати ціаноз і, навіть, синкопальний стан.
Судоми. Спостерігаються у 50-80% випадків і погано піддаються терапії антиконвульсантами. Найчастіше генералізовані тоніко-клонічні приступи, комплексні і прості парціальні судоми.
“Набута” мікроцефалія. Сповільнення темпів росту розмірів голови співпадає з початком захворювання і є наслідком сповільнення росту мозку.
Сколіоз. Він є наслідком дистонії м’язів спини і прогресує по мірі розвитку захворювання.
Дані обстежень:
Практично у всіх випадках, навіть у тих пацієнтів, які не мають клінічно судом спостерігаються зміни на електроенцефалограмі, починаючи з 2-х років. Реєструється збільшення амплітуди і зниження частоти фонового ритму, а також епілептиформні розряди, кількість яких значно більша під час сну. Така комбінація сповільнення фонового ритму після сну і підвищення пароксизмальної активності мозку під час сну значно полегшує діагностику синдрому Ретта і може вважатись його додатковим діагностичним критерієм. Далі характерна для більшості випадків еволюція змін ЕЕГ. З шестирічного віку домінує монотонний альфа ритм, який в подальшому після 20-ти років має тенденцію до локалізації в центро-парієтальній ділянці.
Методи комп’ютерної і магнітно-резонансної томографії, як правило, не дають додаткової інформації про ураження ЦНС. Так, при комп’ютерній томографії відмічається субатрофія кори головного мозку, а магнітно-резонансна томографія виявляє білатеральну атрофію в лобно-скроневих ділянках кори великих півкуль і ознаки атрофії мозочка лише на 3-4 десятилітті життя.
Метод позитронної емісійної комп’ютерної томографії виявляє зниження загального церебрального кровотоку. Найбільше його зниження спостерігається в префронтальній і темпоропарієтальній ділянках великих півкуль, середньому мозку і верхній частині мозкового стовбура, що нагадує поділ кровотоку у дітей грудного віку.
Диференційний діагноз:
При розпізнаванні синдрому Ретта слід врахувати:
- виключні критерії, одного з яких достатньо, щоб відкинути цей синдром;
- додаткові критерії, які можуть бути у хворих, але при цьому ні один не є обов’язковим для встановлення діагнозу;
- необхідні критерії, розроблені Міжнародною асоціацією з вивчення синдрому Ретта.
Слід відмітити, що жіноча стать не входить в їх число, оскільки ведеться пошук і серед хлопчиків. Дані критерії представлені у таблиці:
| Нормальний пренатальний і перинатальний періоди |
| Нормальний обвід голови при народженні з наступним сповільненням росту голови між віком 5 місяців і 4 роки |
| Втрата набутих цілеспрямованих рухів рук у віці від 6-ти до 30-ти місяців, пов’язана в часі з порушенням спілкування |
| Глибоке пошкодження експресивної та імпресивної мови |
| Глибока затримка психомоторного розвитку |
| Стереотипні рухи руками, які з’являються після втрати цілеспрямованих рухів |
| Порушення ходи (атаксія) і забруднення в плануванні дій (апраксія) |
| Дихальні розлади |
| Судоми |
| Спастичність, що поєднується з дистонією і атрофією м’язів |
| Периферичні вазомоторні розлади |
| Сколіоз |
| Затримка росту |
| Гіпотрофічні маленькі ступні |
| Дані ЕЕГ (повільний фоновий ритм в стані бадьорості, епілептифорні розлади переважно під час сну) |
| Затримка внутрішньоутробного розвитку |
| Органомегалія або інші ознаки хвороб накопичення |
| Ретинопатія або атрофія дисків зорових нервів |
| Мікроцефалія при народженні |
| Існування ідентифікованого неврологічного захворювання |
| Набуті внаслідок важкої інфекції або черепно-мозкової травми неврологічні порушення |
Етіологія:
Природа синдрому Ретта генетична. Це спадкове захворювання. Патологія зустрічається майже виключно у дівчаток. Найбільш загальноприйнята гіпотеза про синдром Ретта як Х-зчеплене домінантне захворювання з внутрішньоутробною летальністю у гомозиготних хлопчиків. При цьому майже кожний випадок синдрому є новою мутацією.
На користь Х-зчепленого характеру успадкування свідчить те, що у деяких хворих хромосомні аномалії захоплюють коротке плече хромосоми. У частини хворих була виявлена ламка ділянка в короткому плечі хромосоми Х. У зв’язку з цим передбачають, що ген, відповідальний за розвиток синдрому, знаходиться в короткому плечі хромосоми Х. Однак, незважаючи на інтенсивні молекулярно-генетичні дослідження, спроби картувати ген синдрому Ретта на хромосомі Х не увінчались успіхом.
Запропонована нова гіпотеза стартової пре-мутації, яка може передаватись через ряд поколінь і призвести до повної мутації і синдрому Ретта, як її наслідку. Молекулярно-генетичні пошуки тривають.
Патогенез:
Можливим механізмом розвитку захворювання може бути порушення послідовності процесу реплікації хромосоми Х, яка інактивується. Другий можливий механізм – мітохондріальна дисфункція. Обстеження хворих виявило присутність у них мітохондрій аномальної форми з деструкцією крист і у двох третин хворих, які спостерігались, підвищений рівень молочної та піровиноградної кислот у крові.
Частота виникнення:
1:10000 – 1:15000 дівчаток. Синдром є наступною за частотою після синдрому Дауна специфічною причиною важкої розумової відсталості у дівчаток.
Патоморфологія:
При морфологічних дослідженнях аутопсійного матеріалу померлих хворих виявлено зменшення маси головного мозку на 12-14%, невелике зменшення числа нейронів і гліом в корі великих півкуль і мозочка, передніх рогів спинного мозку і спіральних гангліїв, зниження пігментації substantia nigra. Це вказує на сповільнення розвитку мозку після народження, а до чотирьохрічного віку зупинку його росту, в цілому, і дендритного дерева, зокрема. Спостерігається також сповільнення росту тіла і окремих органів (серця, печінки, нирок, селезінки).
Лікування:
В даний час методи терапевтичної корекції зводяться до симптоматичних засобів. Пропонують дієту з підвищеним вмістом жирів, годування дітей з 3-4-годинними перервами, що призводить до деякої стабілізації стану. При прояві епіприступів призначають антиконвульсанти, хоча ефективність їх незначна. Препаратом вибору є карбамазепін в дозі 10-15 мг/кг. Останнім часом використовують ламотріжин. Для корекції порушень сну пропонується мелатонін.
Один з оптимальних способів корекції рухових розладів – лікувальна фізкультура. Вона допомагає якомога довше зберегти навички ходи. Пропонуються психологічні програми максимального розвитку рухових навичок, які збереглися, і формування на їх основі “мови спілкування”.
Профілактика:
Медико-генетичне консультування.
Пренатальна діагностика:
На даний час не проводиться, так як не відкрито специфічного біологічного маркера синдрому Ретта.
Прогноз:
Остаточно не ясний. Деякі пацієнти гинуть в дитячому і підлітковому віці від дистрофії, ускладнень, пов’язаних з порушенням вентиляції легень внаслідок сколіозу, іноді під час епістатусу. Ряд хворих доживають до 20-30 років. Частина з них нерухомі, приковані до інвалідних візків. Інші мають збережені рухові функції, їх стан залишається відносно стабільним на протязі тривалого часу, іноді, навіть, спостерігається позитивна динаміка в низці симптомів.
Номер з каталогу МІМ:
312750 Rett Syndrome; RTT.
Література:
- Казанцева Л.І., Улас В.Ю. Синдром Ретта у детей (http://www.autism.ru/read.asp?id=77&vol=0).
- Наследственные синдромы и медико – генетическое консультирование /С.И. Козлова, Н.С. Демикова, Е. Семанова, О.Е. Блинникова.- М.: Практика, 1996.- С. 242-243.
- Rett Syndrome. OMIM #312750.
Переглянуто редакційною колегією I.B.I.S.: 15/07/2002
Синдром множинних птеригіумів – зріст дівчат
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”
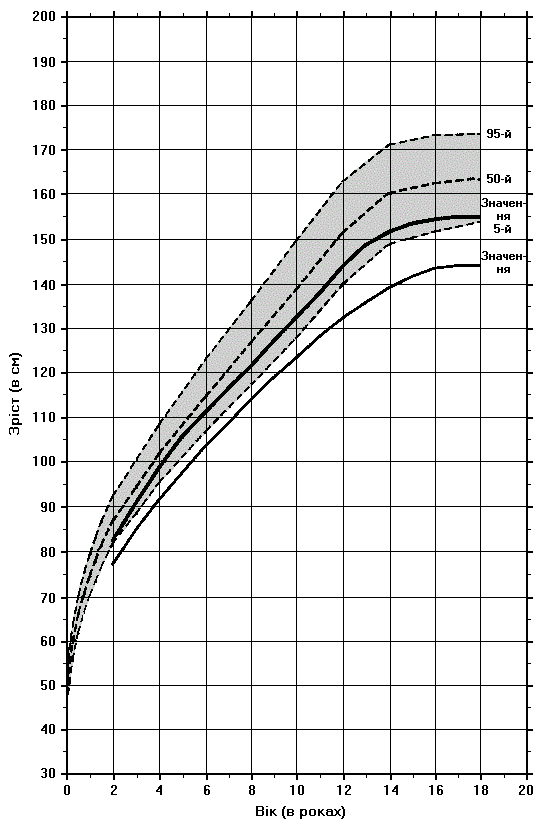
Середній показник для двох підгруп синдрому множинних птерігіумів. Нижня крива представляє “прогресуючу” групу, в якій стан з віком погіршується. Дані отримані в результаті обстеження 13 пацієнтів та порівняння їх з даними про розвиток здорових осіб (зафарбована ділянка). Hall JG: Prog Clin Biol Res 200:155, 1985.
Тератоми пуповини
(Umbilical Teratoma)
Лілія Корольчук
Лікар гінеколог
Рівненського пологового будинку № 2
Визначення:
Тератома – це пухлина, що розвивається з клітин зародка і містить елементи трьох ембріональних шарів.
Частота: невідома.
Ембріогенез:
Зародкові клітини виникають на дорсальній стінці жовточного мішка, звідки вони мігрують до генітального гребеня. Під час цього процесу клітини знаходяться у стінці примітивної кишки. Оскільки пуповина формується в ранні терміни вагітності з випинання примітивної кишки, зародкові клітини можуть потрапляти в пуповину на цій стадії її розвитку. Можливо, що деякі тератоми насправді являють собою акардіальних близнюків і vice verse тератоми.
Патологічна анатомія:
Розміри пухлини можуть перевищувати 9 см в діаметрі. Гістологічно в ній визначаються елементи, які походять з трьох ембріональних шарів. Вона може зазнавати кальцифікації. Тератома може розвиватись в будь-якій ділянці по всій довжині пуповини.
Диференційний діагноз:
Проведення диференційного діагнозу з акардіальним близнюком ґрунтується на тому, що акардіальний близнюк:
- має окрему, хоч і рудиментарну, пуповину;
- не може бути чисто внутрішньопуповинним утвором;
- має деякі організовані частини тіла (наприклад, череп, хребет), тоді як тератома є повністю дезорганізованою масою.
Прогноз:
В одному з трьох описаних випадків плід загинув.
Акушерська тактика:
Діагностика аномалій пуповини, можливо, повинна служити показом до проведення динамічного ехографічного спостереження за ростом плоду і змінами розмірів пухлини, а також до виявлення ознак неімунної водянки. Існує теоретичний ризик компресії судин пуповини утвором, що росте. В таких випадках корисно провести допплерівське дослідження кровотоку в судинах пуповини. У хворих з пухлиною великого розміру пологи рекомендується проводити одразу після підтвердження зрілості легень плоду.
Література:
- Пренатальная диагностика врожденных пороков развития плода /Р. Ромеро, Дж. Пилу, Ф. Дженти, А. Гидини, Дж. С. Хоббинс.- М.: Медицина,1994.- С. 399.
- Benacerraf BR. Ultrasound of Fetal Syndromes. Churchill Livingstone. 1998:351-356.
Переглянуто редакційною колегією I.B.I.S.: 27/08/2003
Синдром Протея
(Proteus Syndrome)
Наталія Олегівна Зимак-Закутня
Зав. медико-генетичною консультацією
Хмельницького клінічного пологового будинку
Синоніми:
Синдром багатоликості, хвороба людини-слона, парціальний гігантизм. Синдром Протея (СП) – рідкісне мультисистемне захворювання із значним клінічним поліморфізмом, що характеризується частковим гігантизмом, ураженням кровоносних та лімфатичних судин. Вперше синдром був описаний Майклом Коеном (Michael Cohen Jr) у 1979 році. У 1983 році німецький педіатр Рудольф Відеман (Rudolf Wiedemann) назвав його синдромом Протея на честь давньогрецького бога Протея-Багатоликого, оскільки чотири хлопчики, у яких був діагностований цей синдром мали різні симптоми і значно відрізнялися один від одного. Часом цей синдром помилково може бути прийнятий за гамартоматозне захворювання. Так, вважалось, що у Джозефа Меррика, відомого як “людина-слон”, був нейрофіброматоз, проте тепер, проаналізувавши все ще раз, клінічні експерти вважають, що фактично це був синдром Протея.
Характерними ознаками СП є:
- шкірні та підшкірні пошкодження;
- неправильний розвиток судин, ліпоми (жирові пухлини), гіперпігментація, декілька типів невусів;
- частковий гігантизм з гіпертрофією кінцівок чи пальців;
- незвичний зовнішній вигляд тіла;
- потовщення ступнів, що нагадують рельєф мозку.
Етіологія та патогенез:
Причиною захворювання є мозаїцизм соматичних клітин за домінантним летальним геном, який поки не ідентифікований. Однак повідомлення про окремі випадки легких проявів захворювання у батьків уражених пробандів ставлять цю гіпотезу під сумнів. Оскільки гіперплазія та гіпоплазія часто виникають разом, іншим можливим поясненням цієї постзиготичної події, є ембріональна соматична рекомбінація, що призводить до щонайменше трьох підвидів клітин – нормальних, гіпертрофічних та атрофічних клітин.
Частота:
Синдром Протея є надзвичайно рідкісним захворюванням, описано близько 200 випадків. Це дає підстави вважати, що поширеність є меншою ніж 1 на 1,000,000 новонароджених.
Смертність/захворюваність:
- Гемігіперплазія (асиметрична гіпертрофія голови, обличчя і пальців) та незначна тканинна гіпертрофія – одні із найсуттєвіших медичних ускладнень.
- Сколіоз або кифоз може бути вираженим і прогресуючим, зумовлюючи респіраторні проблеми. Подовження тулуба та шиї, відносно недорозвинута верхня частина тулуба у поєднанні із гіпертрофією зумовлюють специфічний зовнішній вигляд та супутні функціональні розлади.
- Шкірні ураження викликають серйозні косметичні та функціональні проблеми, оскільки ліпоми, невуси, судинні мальформації можуть мати місцевий інвазивний ріст.
- Часто трапляються кисти легень та внутрішньогрудні або внутрішньочеревні ліпоми.
- Для пацієнтів із синдромом Протея існує високий ризик тромбозу підшкірних вен чи легеневої емболії – серйозних ускладнень перебігу захворювання, навіть в ранньому віці.
- Проблеми із навчанням або затримка розумового розвитку виявляються у частини пацієнтів із синдромом Протея.
Етнічні особливості:
Вибірковості уражень СП за расовою та етнічною належністю не відзначається.
Співвідношення статі:
Ч1 : Ж1. Однак ризик тромботичних ускладнень у чоловіків значно вищий, ніж у жінок.
Вік маніфестації:
Генетичні зміни або соматичні пошкодження клітин, що, очевидно, є причиною синдрому, виникають одразу після зачаття і проявляються у різних клітинних клонах однієї або багатьох ембріональних клітин. Враховуючи значну варіабельність ступеня гіпертрофії та інтенсивності шкірних аномалій, часом встановлення остаточного діагнозу можливе лише у пізньому неонатальному віці чи у ранньому дитинстві.
Клініка:
Існують три загальні критерії, необхідні для встановлення клінічного діагнозу без згадки про специфічні клінічні ознаки:
- мозаїчність уражень;
- прогресуючий перебіг захворювання;
- спорадичність синдрому, тобто він не є спадковим.
Необхідні діагностичні критерії включають наступні категорії:
- Категорія А (1 обов’язково) – невус сполучної тканини.
- Категорія Б (2 обов’язково):
- епідермальний невус;
- диспропорційна гіпертрофія або кінцівок, пальців, черепа, хребців, зовнішнього слухового каналу, селезінки або вилочкової залози;
- двобічна кистаденома яєчників або аденома паротидних слинних залоз у пацієнтів молодших 20 років.
- Категорія В (усі 3 обов’язково):
- ліпоми або фокальна атрофія жирової тканини;
- особливості обличчя: доліхоцефалія, довге обличчя, опущені очні щілини, птоз, сплощене перенісся, вивернуті ніздрі, відкритий рот у стані спокою.
- Асиметрія кінцівок або гіпертрофія черепа можуть бути основними діагностичними критеріями.
- Гіпертрофія кінцівок, пальців або черепа, зумовлена кістковими та м’яко-тканинними змінами.
- Гіперостоз черепа або зовнішнього слухового каналу.
- Типовим є сколіоз, що пов’язаний із непропорційним ростом хребців.
- Поєднання непропорційного росту та фокальної атрофії зумовлює незвичний зовнішній вигляд, що характеризується малою масою м’язів рук, видовженою грудною кліткою, надзвичайно довгою шиєю та м’язевою гіпертрофією стегон.
- Часто зустрічаються кистозні аномалії легень, респіраторні порушення рестриктивного типу внаслідок сколіозу. Рецидивуючі пневмонії, задишка, або непереносимість фізичних навантажень можуть свідчити про значну дихальну недостатність.
- Збільшення органів є менш поширеною ознакою, однак іноді може бути збільшеною селезінка або тимус.
- Невус сполучної тканини є, фактично, патогномонічною ознакою, зазвичай має звивисті контури, локалізується переважно на ступнях.
- Епідермальні невуси гладенькі.
- Ліпоми можуть бути чітко окресленими або мати інвазивний ріст, зумовлюючи внутрішньочеревні та інтраторакальні пошкодження.
- Аномаліям кровоносних та лімфатичних судин властивий прогресуючий характер, на відміну від капілярних гемангіом.
Диференційний діагноз:
- Кліппеля-Треноне-Вебера синдром.
- Нейрофіброматоз.
- Енцефало-краніо-кутанний ліпоматоз.
- Синдром гемігіперплазії-ліпоматозу.
Лабораторні дослідження:
- Молекулярне тестування не проводиться.
- Дослідження тромбоцитів рекомендоване пацієнтам з множинними судинними аномаліями, спленомегалією, особливо при анамнестичних вказівках на петехії та гемангіоми.
- Коагулограма важлива у зв’язку з підвищеним ризиком тромбозу судин.
Інструментальні дослідження:
Рентгенографія:
- детальне дослідження скелету необхідне для підтвердження діагнозу (знімки у передньо-задній та боковій проекціях) у дітей із сколіозом;
- рентгенографія збільшених пальців або кінцівок (коли передбачається ортопедична корекція).
Магнітно-резонансна або комп’ютерна томографія:
- допомагає оцінити внутрішньочерепну анатомію, особливо черепну асиметрію; МРТ голови – необхідна скринінгова процедура для визначення аномалій мозку у дітей із затримкою розумового розвитку;
- тривимірна комп’ютерна томографія інформативна для оцінки гіпертрофії черепа або лицьових структур, однак, ураження і м’яких тканин, і кісток є типовим для подібної гемігіперплазії;
- МРТ грудної клітки, черевної порожнини або кінцівок дозволяє визначити межі підшкірних аномалій (судин або ліпом), що знаходяться глибоко у тканинах, навіть тоді, коли клінічні симптоми відсутні. Недіагностовані вчасно внутрішні ліпоми можуть в подальшому призвести до важких ускладнень;
- комп’ютерна томографія з високою резолюцією грудної клітки інформативна щодо кістозних аномалій легень у пацієнтів з повторними пневмоніями, ателектазом або затрудненим диханням.
Електроенцефалограма призначається усім пацієнтам з судомами в анамнезі.
Спірографічні тести важливі для пацієнтів з дихальними проблемами.
Доплерографія судин для встановлення симптомів глибокого тромбозу судин та легеневого емболії.
Гістологічні дослідження:
Сполучнотканинні невуси мають вигляд щільної колаген-збагаченої тканини. Епідермальні невуси представлені комбінацією гіперкератозу, паракератозу, акантозу, папіломатозу. Ліпоми, як інвазивні так і чітко окреслені, представлені зрілими адіпоцитами.
Лікування:
Лікування передбачає:
- раннє виявлення серйозних медичних проблем;
- застосування профілактичного та симптоматичного лікування.
Гемігіперплазія:
- медичні підходи до лікування є досить обмеженими і повинні визначатись у кожному окремому випадку;
- неоднакова довжина ніг може призвести до подальших ускладнень та корегуватись досвідченим ортопедом;
- збільшені пальці можуть викликати проблеми для пацієнта у повсякденному житті: із письмом, утриманням предметів, харчуванням, одяганням та підбором зручного взуття.
Геміфаціальна макросомія та макроглосія:
- можуть сприяти формуванню неправильного прикусу;
- призначається нарощування зубів, щелепно-лицьова хірургія та постійний нагляд ортодонта.
Сколіоз: раннє встановлення діагнозу запобігає розвитку ускладнень і прогресуванню симптоматики.
Шкірні та підшкірні аномалії: вимагають постійного нагляду, оскільки ліпоми та судинні аномалії можуть призвести не лише до локальних, але і до системних ушкоджень.
Шкірні судинні плями та аномалії: можливе лазерне лікування для видалення шкірних судинних плям та інших аномалій, таких як плями темно-вишневого кольору та капілярні гемангіоми. Видалення плям “кольору кави з молоком” та меланін-пов’язаної гіперпігментації має тимчасовий характер, тому неефективне.
Вторинна тромбоцитопенія (особливо, якщо в анамнезі відзначались гематоми або петехії).
Тромбоз: комплексне лікування призначається пацієнтам, які мають гострий біль у руках та ногах, з видимими рубцями, задишкою або затрудненим диханням.
Внутрішні пошкодження: магнітно-резонансне дослідження грудної клітки та черевної порожнини допомагають встановити внутрішні пошкодження, такі як ліпоми або легеневі кісти.
Медикаментозне лікування не є суттєвим компонентом стандартного лікування синдрому Протея.
Хірургічне лікування:
- До та після оперативного втручання пацієнтам з синдромом Протея необхідно провести лабораторне обстеження зсідання крові.
- Прогресивний сколіоз може вимагати ортопедичного втручання.
- Іноді пацієнти можуть потребувати вкорочення надмірно довгих пальців, щоб носити взуття та користуватись руками.
- Якщо у пацієнта з геміфаціальною макросомією або макроглосією є проблеми з диханням, вигодовуванням або неправильний прикус, необхідне хірургічне втручання за участю щелепно-лицьового хірурга, ортодонта та стоматолога.
- Хоча будь-який досвідчений хірург може видалити великі та небезпечні шкірні та підшкірні плями, консультація пластичного хірурга неодмінна при проведенні операції на обличчі. Підшкірні ураження, які знаходяться на життєво важливих органах, перешкоджаючи зору, або ті, які швидко збільшуються, потребують негайної уваги.
- При потребі хірургічно видаляються внутрішні ліпоми та кисти легень.
Консультування:
- Одним з найголовніших спеціалістів, який повинен постійно оглядати пацієнта з синдромом Протея, звертаючи особливу увагу на розвиток гемігіперплазії та сколіозу, є ортопед.
- Щелепно-лицьовий хірург консультує пацієнтів з черепною асиметрією та геміфаціальною макросомією.
- Консультація загального та пластичного хірурга можлива при необхідності видалення шкірних та підшкірних пошкоджень.
- Консультація нейрохірурга необхідна пацієнтам з ураженнями центральної нервової системи, такими як гіпертрофія головного мозку з або без гідроцефалії.
- Дерматолог може оцінити та спостерігати шкірні і підшкірні ураження, проводити при потребі біопсію.
- Офтальмолог повинен проконсультувати пацієнтів з страбізмом та асиметрією очей.
- Консультація стоматолога може бути необхідною при лікування аномалій зубів та неправильного прикусу.
- Консультація генетика має на меті надання пацієнтові та його родичам додаткової інформації щодо діагностики, тестування та ризику повторення синдрому в наступних поколіннях, планування сім’ї та можливостей пренатальної діагностики.
- Якщо у дитини з синдромом Протея є труднощі у навчанні або затримка розумового розвитку, психіатр та психолог визначають подальшу схему навчання та розвитку дитини. Консультація психолога часто необхідна членам родини, де росте дитина з синдромом Протея.
Враховуючи рідкісність захворювання, “багатоликість” його симптоматики, а також необхідність ретельного постійного нагляду із зосередженням уваги на можливих ускладненнях та його прогресуванні, роль медичного координатора, своєрідного ”диригента” відводиться досвідченому генетику.
Ускладення:
- Гемігіперплазія, асиметрична гіпертрофія кінцівок та пальців та мегалоспондилодисплазія.
- Сколіоз.
- Мікроцефалія, макроглосія, гіперостоз черепа або слухового каналу.
- Невуси сполучної тканини, епідермальні невуси, ліпоми або судинні аномалії.
- Локально інвазивні ліпоми або судинні аномалії.
- Кисти легень.
- Глибокий тромбоз судин, легенева емболія або їх поєднання.
- Кисти яєчників або аденоми привушних слинних залоз.
- Страбізм.
- Аномалії зубів.
- Труднощі з навчанням та затримка розумового розвитку.
Прогноз:
Різноманітні ускладнення, такі як аномалії центральної нервової системи, виражений прогресуючий сколіоз, тромбози, небезпечні внутрішні пошкодження, можуть впливати на тривалість та якість життя пацієнтів з синдромом Протея.
Вчасне встановлення діагнозу та можливих ускладнень, а відтак їх попередження та лікування, значно покращує якість життя та продовжує загальну його тривалість.
Загалом пацієнти з синдромом Протея, які не потребують хірургічного лікування, можуть вести досить активний спосіб життя.
Номер з каталогу МІМ:
176920 Proteus Syndrome.
Література:
- Biesecker LG, Happle R, Mulliken JB: Proteus syndrome: diagnostic criteria, differential diagnosis, and patient evaluation. Am J Med Genet 1999 Jun 11; 84(5): 389-95.
- Biesecker, L. G.; Peters, K. F.; Darling, T. N.; Choyke, P.; Hill, S.; Schimke, N.; Cunningham, M.; Meltzer, P.; Cohen, M. M., Jr.: Clinical differentiation between Proteus syndrome and hemihyperplasia: description of a distinct form of hemihyperplasia. Am. J. Med. Genet. 79: 311-318, 1998.
- Cohen MM Jr: Further diagnostic thoughts about the Elephant Man. Am J Med Genet 1988 Apr; 29(4): 777-82.
- Cohen, M. M., Jr.: Proteus syndrome: clinical evidence for somatic mosaicism and selective review. Am. J. Med. Genet. 47: 645-652, 1993.
- Goodship J, Redfearn A, Milligan D: Transmission of Proteus syndrome from father to son? J Med Genet 1991 Nov; 28(11): 781-5.
- Gorlin JR, Cohen MM, Hennekam RCM. Syndromes of the Head and Neck. Fourth edition. Oxford University Press. 2001:480-484.
- Hamm H: Cutaneous mosaicism of lethal mutations. Am J Med Genet 1999 Aug 6; 85(4): 342-5.
- Happle R: Elattoproteus syndrome: delineation of an inverse form of Proteus syndrome. Am J Med Genet 1999 May 7; 84(1): 25-8.
- Happle R: Lethal genes surviving by mosaicism: a possible explanation for sporadic birth defects involving the skin. J Am Acad Dermatol 1987 Apr; 16(4): 899-906.
- Kruger G, Pelz L, Wiedemann HR: Transmission of Proteus syndrome from mother to son? [letter]. Am J Med Genet 1993 Jan 1; 45(1): 117-8.
- Nazzaro V, Cambiaghi S, Montagnani A: Proteus syndrome. Ultrastructural study of linear verrucous and depigmented nevi. J Am Acad Dermatol 1991 Aug; 25(2 Pt 2): 377-83.
- Plotz SG, Abeck D, Plotz W: Proteus syndrome with widespread portwine stain naevus. Br J Dermatol 1998 Dec; 139(6): 1060-3.
- Rizzo, R.; Pavone, L.; Micali, G.; Nigro, F.; Cohen, M. M., Jr. : Encephalocraniocutaneous lipomatosis, Proteus syndrome, and somatic mosaicism. Am. J. Med. Genet. 47: 653-655, 1993.
- Sigaudy S, Fredouille C, Gambarelli D: Prenatal ultrasonographic findings in Proteus syndrome. Prenat Diagn 1998 Oct; 18(10): 1091-4.
- Smeets, E.; Fryns, J.-P.; Cohen, M. M., Jr.: Regional Proteus syndrome and somatic mosaicism. Am. J. Med. Genet. 51: 29-31, 1994.
- Wiedemann HR, Burgio GR, Aldenhoff P: The proteus syndrome. Partial gigantism of the hands and/or feet, nevi, hemihypertrophy, subcutaneous tumors, macrocephaly or other skull anomalies and possible accelerated growth and visceral affections. Eur J Pediatr 1983 Mar; 140(1): 5-12.
Переглянуто редакційною колегією I.B.I.S.: 03/02/2004
Синдром множинних птеригіумів – зріст хлопчиків
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”

Середній показник для двох підгруп синдрому множинних птерігіумів. Нижня крива представляє “прогресуючу” групу, в якій стан з віком погіршується, особливо у хлопчиків. Дані отримані в результаті обстеження 13 пацієнтів та порівняння їх з даними про розвиток здорових осіб (зафарбована ділянка). Hall JG: Prog Clin Biol Res 200:155, 1985.
Пробошизис
(Proboscis)
Лілія Георгіївна Романенко
Лікар-гінеколог
пологового будинку № 1, м. Рівне
Визначення:
Пробошизисом називають хоботоподібний відросток з одним чи двома внутрішніми отворами, зазвичай у поєднанні з відсутністю носа.
Пробошизис майже завжди поєднується з голопрозенцефалією. Припускається, що в таких випадках первинне ураження прехордальної мезенхіми призводить до патологічного розвитку серединних структур обличчя. Внаслідок патологічного розвитку в носовій ділянці може виникнути зрощення (злиття) нюхальних плакод і формування пробошизиса. Порушення морфогенезу серединних структур обличчя може призвести до різного розміщення пробошизиса по відношенню до очей.
Діагностичні критерії:
Виявлення хоботоподібної структури зазвичай з єдиним центральним отвором на місці носа чи між очними ямками.
Клінічна характеристика. Асоційовані аномалії:
Пробошизис, зазвичай, має один центральний отвір. Він може локалізуватись над орбітою (циклопія, етмоцефалія) чи у місці звичайної локалізації носа, між орбітами (цебоцефалія). Отвір пробошизиса не з’єднується з хоанами. Решітчаста кістка, носові раковини, носова і слізна кістки відсутні. Типова відсутність розщілини губи і піднебіння при циклопії, етмоцефалії і цебоцефалії. Повідомлення про пробошизис при відсутності голопрозенцефалії зустрічаються дуже рідко. В таких випадках, зазвичай, відмічають односторонню аплазію носа, а пробошизис виявляють на місці відсутніх носових структур. В рідких випадках пробошизис буває двобічний.
Частота:
Частота аномалії невідома.
Прогноз і акушерська тактика:
В більшості випадків аномалія поєднується з голопрозенцефалією. Необхідно дослідити каріотип плода.
Література:
- Пренатальная диагностика врожденных пороков развития плода /Р. Ромеро, Дж. Пилу, Ф. Дженти, А. Гидини, Дж.С. Хоббинс.- М.: Медицина, 1994.- C. 104-105.
- Aase JM. Diagnostic Dysmorphology. Plenum Medical Book Company. New York and London 1990:91.
- Gorlin RJ, Cohen MM, Hennekam RCM. Syndromes of the Head and Neck. Oxford University Press 2001:715, 719.
- Warkany J. Congenital Malformations. Notes and Comments. Year Book Publisher. Chicago 1981:203-204, 589.
Переглянуто редакційною колегією I.B.I.S.: 05/01/2004
|
|
|
|
|
|





