
Serhiy
Кампомелічна дисплазія
(Campomelic Dysplasia)
З.О. Сосинюк
Лікар УЗД
Рівненського клінічного лікувально-діагностичного центру
Г.І. Білякова
Лікар акушер–гінеколог
Рівненського клінічного лікувально-діагностичного центру
Включення:
Кампомелічна карликовість, кампомелічний синдром, СМД–1.
Визначення:
Кампомелічна дисплазія відноситься до групи остеохондродисплазій, які призводять до загибелі у внутрішньоутробному або неонатальному періодах. Термін “кампомелія” означає викривлення кінцівок.
Кампомелічна дисплазія являє собою патологічний стан, який характеризується викривленням довгих трубчастих кісток, особливо ніг, гіпоплазією лопаткової кістки, пласким обличчям.
Частота:
За даними різних авторів частота кампомелічної дисплазії коливається в середньому від 0,05 до 0,16 випадків на 10000 пологів.
Основні діагностичні критерії:
Найбільш типова і незмінна ознака – кампомелія, яка характеризується викривленням кісток гомілки, рідше – стегнових кісток. Трубчасті кістки нормальної довжини або вкорочені. Черепно–лицеві аномалії, наприклад, макроцефалія, пласке обличчя, низько розташовані вушні раковини, запале перенісся, гіпертелоризм, мікрогнатія, глоссоптоз, розщілина піднебіння зустрічаються у 90-99% випадків. Гіпоплазія лопаткової кістки визначається у 92% хворих. Грудна клітка вузька, дзвоноподібна. При рентгенографії визначають сколіоз, короткі і тонкі ключиці, відсутність мінералізації грудини, гіпопластичні тіла і відростки хребців, недостатня осифікація проксимальних та дистальних епіфізів гомілки, стегон, таранних кісток. Крила клубових кісток вертикально звужені у 98% хворих, відносно широкий тазовий отвір. Часто зустрічається вивих стегон, еквіноварусна установка ступнів. Характерна м’язова гіпотонія.
Поєднані аномалії:
Гідроцефалія зустрічається у 23% хворих, описані аріненцефалія, ліссенцефалія. Вроджені вади серця (дефекти міжпередсердної та міжшлуночкової перетинок, тетрада Фалло, стеноз аорти) – у 30% хворих, гідронефроз (частіше однобічний) – у 30% хворих, зустрічається гіпоплазія або полікистоз нирок, гіпоплазія легень, зниження слуху. Гермафродитизм, чоловічий каріотип у хворих з жіночим фенотипом, гонади представлені яєчками, зустрічаються у 50% випадків.
Пренатальна діагностика:
Кампомелічна дисплазія пренатально діагностується на основі виявлення затримки внутрішньоутробного розвитку в поєднанні з патогномонічною ознакою –викривленням кісток гомілки, іноді стегон. Грудна клітка – вузька, у формі дзвону. Виявляють поєднані аномалії – гідроцефалію, вади серця, гідронефроз, розщілину піднебіння. Як і інші скелетні дисплазії, кампомелічна дисплазія може поєднуватись з багатовіддям, яке нерідко супроводжується передчасними пологами. Найбільш рання пренатальна діагностика може бути здійснена в 17-20 тижнів вагітності.
Диференційний діагноз:
Недосконалий остеогенез, гіпофосфатазія, танатоформна дисплазія, мезомелічна дисплазія (тип Рейнхарда). Крім того, вроджене викривлення трубчастих кісток може бути доброякісним станом, що не супроводжується іншими метаболічними порушеннями.
Генетичне консультування:
Мутації в SOX9, статево-визначений район Y(SRY) сумісний ген, що розміщений в 17q24 спричиняє і кампомелічну дисплазію, і зміну статі. Тип успадкування – аутосомно-домінантний.
Прогноз:
Ніхто із хворих, зазвичай, не доживає до ювенільного віку. Діти переважно помирають в неонатальному періоді. Основна причина смерті – дихальна недостатність. У тих, що вижили, виявляють втрату слуху, виражену затримку психомоторного розвитку, кіфосколіоз.
Співвідношення статей: Ч < Ж.
Вік маніфестації: пренатальний період.
Акушерська тактика:
Переривання вагітності пропонують до досягнення періоду життєздатності плоду. У пізні терміни вагітності слід проводити динамічне ехографічне спостереження для виключення багатовіддя. У випадку загрози передчасних пологів призначення токолітиків невиправдане. Родорозрішення необхідно проводити через природні родові шляхи.
Номер з каталогу МІМ:
114290 Campomelic Dysplasia
Література:
- Наследственные синдромы и медико–генетическое консультирование /С.И. Козлова, Н.С. Демикова, Е. Семанова, О.Е. Блинникова.- М.: Практика, 1996.- C. 107-108.
- Пренатальная диагностика врожденных пороков развития плода /Р. Ромеро, Дж. Пилу, Ф. Дженти, А. Гидини, Дж.С. Хоббинс.- М.: Медицина, 1994.- C. 349-351.
- Тератология человека /Под ред. Г.И. Лазюка.- М.: Медицина, 1979.- C. 357-359.
- Cremin BJ, Orsmond G, Beighton P. Autosomal recessive inheritance in camptomelic dwarfism (Letter). Lancet 1973;I:488-489.
- Fontaine G, Farriaux JP, Tilmont P, Peuzin F, Delecour M. Le conseil genetique dans la dysplasie campomelique (a propos de deux observations). J Genet Hum 1980;28:267-279.
- Hall B, Spranger JW. Campomelic dysplasia: further elucidation of a distinct entity. Am J Dis Child 1980;134:285-289.
- Hovmoller ML, Osuna A, Eklof O, Fredga K, Hjerpe A, Lindsten J, Ritzen M, Stanescu V, Svenningsen N. Camptomelic dwarfism. A genetic determined mesenchymal disorder combined with sex reversal. Hereditas 1977;86:51-62.
- Jones K.L. Smith’s recognizable Patterns of Human Malformation. Philadelphia: W.B.Saunders Company 1997:344-346.
- Warkany J. Congenital Malformations. Notes and Comments. Chicago: Year Book Publisher 1981;768-782.
Переглянуто редакційною колегією I.B.I.S.: 11/03/2002
Діастрофічна дисплазія – зріст
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”.
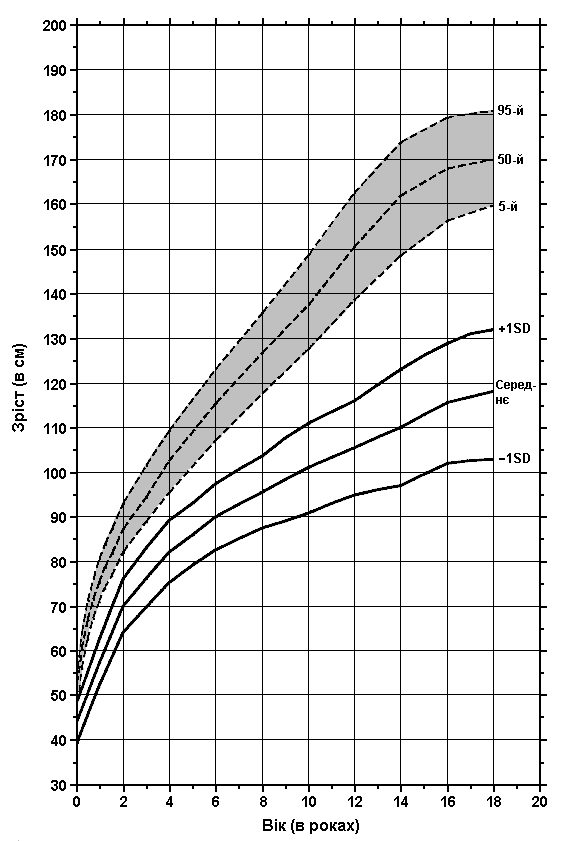
Зріст осіб з діастрофічною дисплазією. Дані отримані в результаті обстеження 72 пацієнтів (38 хлопчиків та 34 дівчинки). Ці дані порівнюються з розвитком здорових осіб (зафарбовані ділянки), які так само складаються з інформації про осіб обох статей. Horton WA et al: Am J Dis Child 136:136, 1982.
Діастрофічна дисплазія
(Diastrophic Dysplasia)
Л.О. Ярмолюк
Лікар відділення ультразвукової діагностики
Рівненської обласної клінічної лікарні
Включення:
Синдром діастрофічного нанізму, діастрофічна карликовість. Термін “діастрофічна” означає скручена, викривлена і пов’язаний з типовим зовнішнім виглядом хворих.
Основні діагностичні критерії:
Діастрофічна дисплазія характеризується мікромелією, клишоногістю, деформацією китиць, множинними згинальними контрактурами суглобів, сколіозом.
Етіологія:
Ген розміщений на дистальній частині довгого плеча хромосоми 5. Послаблення його функціонування призводить до продукування несульфатних протеогліканів в хрящовому матриксі, що і вважається головною причиною цього порушення. Захворювання передається за аутосомно-рецесивним типом.
Клінічна картина:
Захворювання розпізнається при народженні, але легкі форми можуть діагностуватися і в більш пізньому віці. До клінічних ознак, що виявляються при народженні, відносяться малий зріст, мікромелія (переважно ризомелічного типу), множинні згинальні контрактури суглобів, особливо великих, деформація китиць (проявляється короткими і широко розставленими пальцями, відведеним великим пальцем – “рука, що хапає”) і виражена еквіно-варусна деформація ступні. Мозкова частина черепа не змінена. Часто відмічають мікрогнатію і розщілину піднебіння, обмежене розгинання в ліктьових суглобах.
З часом у дитини прогресує кіфосколіоз (49% у дівчат і 22% у хлопців) з можливим порушенням дихання, закрита спинно-мозкова кила від С3-С4 до верхніх грудних хребців (73% у дівчат і 59% у хлопчиків). Хода відрізняється характерними вихиляючими рухами внаслідок вивиху стегна та геноварусною деформацією колінного суглобу. Запалення зовнішнього вуха призводить до типової деформації вушних раковин, відомої під назвою вуха у вигляді “кольорової капусти” (84% пацієнтів).
Діастрофічна дисплазія – захворювання з генералізованим пошкодженням хрящової тканини. У хондроцитах та хрящовому матриксі відбуваються дистрофічні процеси з наступним утворенням фіброзної рубцевої тканини та оссифікацією. Ці процеси стають причиною контрактур. Пластинки росту також пошкоджені. Відомих на сучасному етапі біохімічних пошкоджень не виявлено.
Поєднані аномалії:
Латеральне зміщення надколінника. Гіпоплазія шийних хребців. Неповний вивих шийних хребців, вивих ліктьового суглобу, гіпереластичність шкіри, крипторхізм. Рання мінералізація ребер, інтракраніальна кальцифікація. Глухота, вторинна до зрощення кісточок, стеноз зовнішнього вушного каналу. Ларингеальний стеноз. Капілярна гемангіома середньої лінії обличчя.
Пренатальна діагностика:
Пренатальна діагностика можлива у плодів високого ризику. Спектр прояву ознак захворювання широкий, тому в деяких випадках воно не може бути діагностоване пренатально.
Диференційний діагноз:
Крім вкорочення кінцівок найбільш характерною ознакою діастрофічної дисплазії бувають множинні контрактури ніг і рук. Але вони зустрічаються і при інших захворюваннях, таких як множинний вроджений артрогрипоз, мезомелічна дисплазія типу Нівергельта. Однією з скелетних дисплазій, з якою необхідно проводити диференційний аналіз, є синдром Вайсенбахера-Цваймюллера, який проявляється вкороченням кінцівок, мікрогнатією, дефектами лицевого скелету (розщілина піднебіння, гіпертелоризм, запале перенісся). При цьому захворюванні контрактури відсутні.
Прогноз:
Смертність в періоді новонародженості висока. Основними її причинами є дихальна недостатність та аспіраційна пневмонія. Загальний стан здоров’я у тих, хто вижив, зазвичай, добрий. Інтелект хворого збережений. Прогресуючий кіфосколіоз і артропатія призводять до вираженого порушення фізичного розвитку, а при тяжких формах – до дихальної недостатності, до стиснення спинного мозку. Потрібно завжди зважати на можливість атлантовісьової нестабільності. Зріст дорослої людини коливається від 100 до 140 см, в середньому – 125 см.
Акушерська тактика:
До досягнення періоду життєздатності плоду може бути запропоноване переривання вагітності. Після настання цього періоду стандартна акушерська тактика не змінюється.
Номер з каталогу МІМ:
222600 Diastrophic Dysplasia; DTD
Література:
- Козлова С.И., Демикова Н.С., Семанова Е., Блинникова О.Е. Наследственные синдромы и медико-генетическое консультирование. М.: Практика, 1996.- С. 97-98.
- Рожеро Р., Пилу Дж. Пренатальная диагностика врожденных пороков развития плода.- М.: Медицина, 1994.- С. 363-366, 353-355.
- Jones KL. Smith’s recognizable patterns of human malformations. 5th edition. Philadelphia: W.B. Sauders Company, 197б:376-378.
- Warkany J. Congenital malformations: Notes and comments. Chicago, 1981:801-803.
Переглянуто редакційною колегією I.B.I.S.: 11/02/2002
Дивіться також:
Мандібулофаціальний дизостоз
(Mandibulofacial Dysostosis)
Л.М. Зайцева
Лікар-неонатолог
Волинського обласного дитячого територіального медичного об’єднання
Синоніми:
- синдром Трічера-Коллінза;
- Франческетті-Клейна синдром;
- синдром Томсона;
- нижньощелепно-лицевий дизостоз.
Мінімальні діагностичні ознаки:
- антимонголоїдний розріз очей;
- гіпоплазія виличних кісток;
- колобоми нижніх повік;
- аномалії зовнішнього вуха.
Частота виникнення: 1:50000 новонароджених.
Синдром вперше описаний Томсоном в 1846-1847 роках, Трічер Коллінз описав обов’язкові компоненти цього синдрому, Франческетті і автори в 1940 році запропонували термін мандібулофаціальний дизостоз.
Патогенез:
Мадібулофаціальний дизостоз – вроджений дефект голови та обличчя, який виник внаслідок порушення краніо-фаціального розвитку.Він включає зміни структур, що походять з першої та другої горлової дуги, щілини та кишені. Ген кодує синтез ядерного фосфопротеїну. Мутації призводять до передчасного закінчення кодування.
Генне картування:
Ген цього синдрома (Treacle або ТСОF1) картується на 5q32-33,1. В легких випадках асоціюється з інтерстіціальною делецією, дел. (3) (р23; р24).
Тип успадкування:
Аутосомно-домінантний з різною експресивністю та високою пенетрантністю, можливий аутосомно-рецесивний при гонадоідальному мозаїцизмі переважно у старших батьків. Допускають, що важкі випадки є результатом уніпарієтальної дисомії, яка приводить до гомозиготності за цим геном. В 60% виявляються нові мутації.
Клінічна характеристика:
- антимонголоїдний розріз очей (89%);
- двобічна гіпоплазія виличних кісток (81%);
- колобоми нижніх повік (69%);
- гіпоплазія нижньої щелепи (78%);
- аномалії вушних раковин;
- “пташине” обличчя.
Часто зустрічається атрезія зовнішнього слухового каналу (36%), провідникова глухота (40%), відсутність вій на нижній повіці (53%), високе піднебіння або розщелина його (35%), макростомія, відкритий прикус, в деяких випадках – ріст волосся на щоках (26%), атрезія хоан, преаурікулярні вирости або фістули, відсутність білявушних слинних залоз, мікрофтальмія, колобоми верхньої повіки, райдужки, екзофтальм, вади серця, скелетні аномалії, крипторхізм.
Кістки черепа: склепіння черепа є практично нормальним, але радіографічне обстеження виявляє недорозвиток супраорбітального гребеня. Виличні кістки відсутні, але частіше недорозвинені. Скулові відростки лобної кістки гіпоплазовані. Мастоїдальний відросток непневматизований і часто склерозований. Параназальні синуси маленькі і можуть бути відсутні. Характерний гіпертелоризм. Нижній край орбіти з дефектами, інтраорбітальний утвір може бути відсутній. Основа черепа кіфотична. Мандібулярний і коронарний відростки щелепи гіпоплазовані, плоскі або аплазовані, поверхня тіла нижньої щелепи вигнута, кут – тупий або нормальний, гілки недорозвинуті, відростки вкриті гіаліновим хрящем. Відростки шийних хребців – короткі.
Очі: очні розрізи короткі і скошені донизу, колобоми зовнішньої третини нижньої повіки. У половини пацієнтів відсутні вії медіальніше дефекта, можлива колобома райдужки, характерна атрезія сльозних протоків, відсутні білявушні слюнні залози.
Вуха: зовнішні вушні раковини деформовані, зміщені донизу, у однієї третини – зарощений зовнішній слуховий канал, у 60% – мікротія, слухові кісточки, кохлеарний та вестибулярний апарат, середнє вухо та епітимпанічний простір можуть бути відсутні або недорозвинені, внутрішнє вухо є нормальним, характерні також преаурікулярні вирости або фістули.
Ніс: тонкий, дзьобоподібний, тому що хрящі гіпоплазовані. Описана атрезія хоан.
Розумовий статус: інтелект – нормальний, розумова відсталість розглядається, як “вторинна”, внаслідок втрати слуху.
Аномалії ротової порожнини: у 35% – розщілина піднебіння, макростомія у 15%, вроджена палатофарингеальна недостатність у 34%, піднімаючі м’язи губи недорозвинуті, глоткова гіпоплазія може привести до смерті у періоді новонародженості.
Диференціальна діагностика:
- акрофаціальний дизостоз;
- геміфаціальна мікросомія;
- Гольденхара синдром;
- Синдром Дауна.
Пренатальна діагностика:
УЗД плода в другому триместрі і в 20 тижнів гестації, фетоскопія.
Лікування:
Постійне спостереження та надання медичної допомоги протягом перших п’яти років життя:
- трахеостомія при затрудненні дихання, накладення гастростоми для необхідного повноцінного харчування;
- пластичні операції з метою корекції верхньої та нижньої щелепи;
- ортодонтичне лікування аномального прикуса;
- при наявності колобоми реконструкція нижньої повіки;
- протезування вушних раковин та застосування “штучного вуха” або BAHA (імплантованого в кістку за допомогою титанового фіксатора звукового процесора, який можна приєднувати або від’єднувати по потребі).
Прогноз:
Враховуючи нормальний інтелект при ранній реабілітації слуху можливий нормальний розумовий розвиток. Проблеми дихання та вигодовування з віком зникають. Пластична хірургія дає можливість пацієнтам нормально розвиватись, рости і бути соціально адаптованими.
Номер з каталогу МІМ:
- 154500 Treacher Collins-Franceschetti Syndrome; TCOF.
- 248390 Mandibulofacial Dysostosis, Treacher Collins Type, Autosomal Recessive.
Література:
- Herring SW, Rowlatt UF, Pruzansky S. Anatomical abnormalities in mandibulofacial dysostosis. Am J Med Genet. 1979;3(3):225-9.
- Sulik KK, Johnston MC, Smiley SJ, Speight HS, Jarvis BE. Mandibulofacial dysostosis (Treacher Collins syndrome): a new proposal for its pathogenesis. Am J Med Genet. 1987 Jun;27(2):359-72.
- Pron G, Galloway C, Armstrong D, Posnick J. Ear malformation and hearing loss in patients with Treacher Collins syndrome. Cleft Palate Craniofac J. 1993 Jan;30(1):97-103.
- Wise CA, Chiang LC, Paznekas WA, Sharma M, Musy MM, Ashley JA, Lovett M, Jabs EW. TCOF1 gene encodes a putative nucleolar phosphoprotein that exhibits mutations in Treacher Collins Syndrome throughout its coding region. Proc Natl Acad Sci U S A. 1997 Apr 1;94(7):3110-5.
Переглянуто редакційною колегією I.B.I.S.: 27/08/2003
Дивертикул Меккеля
(Meckel Diverticulum)
О.О. Михасюк
Спеціаліст з інформаційного забезпечення
Українсько-Американської Програми запобігання вродженим вадам розвитку
Опис:
Дивертикул – це випинання стінки порожнистого органу (сечового міхура або кишківника). Дивертикул Меккеля (залишок жовчного стовбура – утвір, що іноді зустрічається в нижній третині здухвинної кишки і є залишком не повністю редукованої судини, яка зв’язувала ембріон з жовточним мішком на ранніх стадіях розвитку) являє собою пальцеподібне випинання стінки кишки, що потребує лікування. Він може запалюватися та кровоточити, викликати кишкову непрохідність або інвагінацію. В таких випадках необхідним є хірургічне втручання, зазвичай невідкладне, через загрозу ускладнень, пов’язаних з дивертикулом Меккеля.
Ця вроджена патологія є досить поширеною і зустрічається приблизно в 1% людей, але її ускладнення розвиваються лише в деяких дітей. Дивертикул Меккеля не є спадковою аномалією та не залежить від генетичного дефекту. Він виникає у випадках, коли протока, що є необхідною лише протягом першого місяця розвитку плоду, зберігається протягом всього ембріонального періоду до самого народження. Іноді дивертикул Меккеля залишається прикріпленим до пупка, так, як це було до народження, утворюючи невеликий, частково рухомий тканинний місток між стінкою живота та кишечником.
Проблеми, пов’язані з дивертикулом Меккеля, можуть виникати в будь-якому віці, але майже половина випадків ускладнень відбувається серед дітей дворічного віку. 4 з 5 дітей, яким проводяться оперативні втручання з приводу ускладнень дивертикула Меккеля є молодшими 10 років. Співвідношення статей приблизно однакові – майже 1:1 (спостерігається незначне переважання хлопчиків). Переважна більшість ускладнень пов’язана з кровотечею з дивертикула.
Причини:
Якщо дивертикул Меккеля вкритий зсередини слизовою оболонкою, яка виділяє такі ж кислі травні соки, як і шлунок, може розвинутись виразка, що кровоточить. Зазвичай вона розташована в місці відкриття дивертикулу в петлю кишечника.
Ознаки і симптоми:
Загальною ознакою дивертикулу Меккеля є раптова кровотеча з прямої кишки, що супроводжується різким болем, або криваві випорожнення. Кровотеча може як раптово розпочатися, так і раптово припинитися. Але часом кровотеча є настільки важкою, що дитині може загрожувати втрата крові в разі ненадання своєчасної медичної допомоги. Часто розвивається запалення, яке супроводжується нудотою та блювотою.
Слід уважно спостерігати за клінічними проявами, оскільки часто симптоми дивертикула Меккеля нагадують ознаки гострого апендициту.
Ускладнення:
Непрохідність кишківника може розвинутись, якщо дивертикул вивернеться назовні (направиться до внутрішнього сегменту кишки), викликаючи інвагінацію. Оскільки потік крові до сегменту, що був поглинутий, зменшується, тканинам бракує кисню і може розвинутися гангрена. Це пов’язано з відмиранням тканин та виникненням інфекції. Крім того, непрохідність може розвинутись, якщо петля кишки починає закручуватися навколо тканинного містка до пупка. Біль, що викликається непрохідністю кишківника, відрізняється від болю, що асоціюється з виразками тонкого кишківника або шлунка. Цей біль не можна зняти засобами для зниження кислотності, вживанням рідини або їжі. Крім того, біль при непрохідності зазвичай чітко обмежується певною ділянкою живота. Також існує вірогідність попадання в дивертикул Меккеля їжі або сторонніх предметів, що, в свою чергу, може викликати запалення. Якщо розрив дивертикулу відбувається у дитини до 2 років, шансів на успішну операцію та одужання є менше, ніж в більш дорослих дітей.
Діагноз:
Будь-яка дитина з кривавими випорожненнями або яскравою чи темно-червоною кровотечею з прямої кишки має бути негайно обстежена лікарем. Після з’ясування симптомів захворювання лікар повинен провести ретельний фізичний огляд. Також в хворого беруться аналізи крові для виявлення можливої інфекції. УЗД або сканування живота можуть допомогти точніше встановити розташування джерела кровотечі.
Лікування:
Одразу після встановлення діагнозу дитині необхідно провести термінову операцію. Інколи необхідним є також переливання крові. Хірургічне втручання полягає в видаленні дивертикулу, іноді з сегментами кишки, що оточують ділянку запалення. Після цього два здорових кінця кишки зшивають. Зазвичай одужання є повним та без ускладнень. Проте, якщо операція не була проведена до того, як розвинулись ускладнення, вірогідність виживання та повного одужання пацієнта залежать від ступеня важкості ускладнень та стану здоров’я дитини до хвороби.
Профілактика:
Методів запобігання дивертикулу Меккеля – не існує.
Переглянуто редакційною колегією I.B.I.S.: 27/11/2002
Дивіться також:
- Дивертикул Меккеля (інформація для спеціалістів)
Дивертикул Меккеля
(Meckel Diverticulum)
Т.Є. Голуб
Лікар-неонатолог
Дитяча обласна лікарня, м. Рівне
Визначення:
Дивертикул Меккеля – один з різновидів персистенції жовточної протоки.
Патогенез:
Необлітерованою залишається жовточна або пупочно-брижова протока. Знаходиться на протилежному брижі боці клубової кишки, на відстані 20-70 см від ілеоцекального клапана. По формі нагадує короткий червоподібний відросток. Частіше має конічну або циліндричну форму. Його розміри становлять 2-5 см.
При гістологічному дослідженні стінки Меккелевого дивертикулу в низці випадків знаходять дистоловану слизову оболонку шлунка або різних відділів кишківника. Рідше спостерігається тканина підшлункової залози. Дистопія атипової залозистої тканини є причиною одного з ускладнень Меккелевого дивертикулу – ерозії його стінки і кишкової кровотечі.
Клініка:
Частіше виявляють випадково при лапаротомії, що проводиться з іншого приводу або в зв’язку з розвитком ускладнень. Дуже рідко жовточна протока утворює фістулу між клубовою кишкою та пупком, через яку виділяються калові маси.
Діагностика:
Діагностується важко. Про дивертикул Меккеля частіше думають у випадку рецидивуючих кишкових кровотеч. Рентгенологічне дослідження безрезультатне. Для кінцевого виключення діагнозу застосовують пробну лапаротомію.
Ускладнення:
- Кишкова кровотеча – виникає гостро. Кров в калі темно-коричневого кольору. При масивній кровотечі швидко розвивається анемія. Кровотеча може повторюватися неодноразово.
- Дивертикуліт – перебігає з симптомами, подібними до гострого апендициту (нудота, блювота, болі в животі, підвищення температури тіла, лейкоцитоз). Відрізнити ці захворювання практично неможливо. Тому при відсутності змін в червоподібному відростку під час лапаротомії необхідно провести ревізію тонкої кишки на відстані близько 70 см. В запізнілих випадках діагностики дивертикулу наступає перфорація і розвивається перитоніт.
- Кишкова інвагінація – починається з дивертикуліту, перебігає з типовими симптомами (різкий початок, нападоподібні болі в животі, блювота, кишкова кровотеча). Дивертикул виявляється на операції після дезінвагінації.
- Кишкова непрохідність – може викликатися перекрученням кишкових петель навколо дивертикулу. Клінічна картина типова для кишкової непрохідності.
Частота: 2-3 % серед новонароджених.
Співвідношення статей:
Ж:Ч = 1:3. Проте деякі дослідження показують, що Ж:Ч = 1:1, але ускладнення, виникають частіше у хлопчиків.
Лікування:
Оперативне видалення Меккелевого дивертикулу.
Прогноз:
При своєчасному оперативному втручанні у випадку виникнення ускладнень, сприятливий.
Поєднані симптоми:
Дивертикул Меккеля поєднується з іншими ВВР – мікроцефалією, spina bifida, вродженими вадами серця, аномаліями селезінки, нирок та сечоводу. При патанатомічному дослідженні дітей з Трисомією 18 дивертикул Меккеля виявлений в більше, як 20% випадків. При трисомії 13-15 – в 10% випадків.
Номер з каталогу МІМ:
155140 Meckel Diverticulum.
Літературні джерела:
- Баиров Г.А. Срочная хирургия детей (руководство для врача).- СПб: Питер, 1997.- C. 211-219.
- Лазюк Г.И. Тератология человека. М.: Медицина, 1979.
- Шабалов Н.П. Детские болезни. СПб: Питер, 1999.
- Warkany L, Passarge E, Smith LB. Congenital malformations in autosomal trisomy syndrome. Am.J.Dis.Child. 112:502;1066.
Переглянуто редакційною колегією I.B.I.S.: 02/02/2001
Дивіться також:
- Дивертикул Меккеля (інформація для батьків)
Клишоногість та інші деформації стопи
(Clubfoot)
І.Є. Харитонова
Координатор Українського Альянсу
із запобігання вродженим вадам розвитку
Клишоногість є однією з головних ортопедичних (хвороби кісток та суглобів) проблем дітей вже понад 200 років, відколи лікарі почали займатися нею. Це є одна з найбільш поширених вад розвитку. У Сполучених Штатах щороку біля 5000 немовлят (один на 735) страждають над тяжкі форми клишоногості. Хлопчики вражаються на тяжкі форми клишоногості вдвічі частіше за дівчаток. Легкі форми – більш поширені, співвідношенність між статтю не відмічається.
Що таке клишоногість?
Слово “клишоногість” використовується для визначення певних аномалій щиколотки та стопи, які звичайно існують з народження. Дефект може бути у легкій чи більш тяжкій формі, може бути ураженою одна кінцівка чи обидві. У медичній термінології слову “клишоногість” відповідає термін “еквіноварусна деформація стопи”. Стопа скручена усередену і униз. Якщо обидві стопи “чепляються”, то пальці однієї ноги повернуті до пальців другої замість того, щоб бути паралельно. Зв’язка п’яти досить часто буває надто короткою, що робить неможливим без допомоги спеціалістів повернути стопу у нормальне положення догори.
Які ще вади стопи поширені?
Найбільш поширеним типом вад стопи є еквіноварусна деформація стопи. Вона призводить до того, що стопа стає піднятою під гострим кутом до п’яти, спрямована вгору і назовні. У багатьох випадках стопа своєю верхівкою може торкатися гомілки. Легка форма цієї деформації звичайно минає без лікування і ніяких ушкоджень не лишається.
Аддукція (приведення) плесна є іншою легкою формою деформації стопи за якої передня частина стопи вивернута усередину. Не дивлячись на те, що ця вада існує з народження, вона може лишатися невиявленою декілька перших місяців від народження дитини. Ця деформація примушує дитину мати ходу пальцями усередину (загребати ногами). Навіть тяжкі випадки не потребують лікування, бо цей стан виправляється сам по собі. Однак найбільш тяжкі випадки лікуються, щоб допомогти стопі краще рухатись і щоб запобігти подальших проблем з взуттям.
Яких страджань завдає клишоногість (еквіноварусна деформація стопи) дитини?
Клишоногість не завдає болю і не турбує дитину, доки він чи вона не починає ставати на ніжки та ходити. Якщо клишоногість не лікувати, формується незграбна хода, бо щиколотка лишається покрученою і стопа не може рухатись вгору і вниз як це має бути за норми.
У разі ураження обох стоп (як це і буває насправді у 50% випадків), дитина ходить на підйомі своду стопи, або якщо стопа сильно викручена, на бокових поверхнях чи буває на верхній поверхні стопи замість ступні. Частина, якою користуються для ходьби може інфікуватися, на ній можуть утворюватись великі і болісні мозолі. а нога в цілому часто не може зростати як це має бути. Також утворюються болісні зміни у суглобах.
В разі ураження однієї стопи, ця стопа та литка будуть меньші за здорову.
Що є причиною клишоногості?
У більшості випадків причина виникнення невідома. В минулому були такі думки, що стопи дитини покручені чи зведені судомами через положення, у якому знаходилось дитя у материнському лоні. Це є відповідним у тих випадках деформацій стопи, що самі виправляються після народження, еквіноварусна деформація стопи і аддукція плесни, включно. Багато вчених вважають, що клишоногість (еквіноварусна деформація стопи) формується ще на ранніх етапах вагітності, можливо десь на 10-12 тижні гестації.
Клишоногість спричинена поєднанням спадкових та інших чинників, що можуть впливати на пренатальне формування, а саме, інфекції, наркотики/ліки, захворювання чи інші фактори внутрішньоутробні чи навколишнього середовища. Незважаючи на це, у більшості дітей з клишоногістю немає інших вроджених вад розвитку, зрідка можуть траплятися інші вади. В невеликій частці випадків клишоногість утворюється як частина синдрому, до якого входять декілька вроджених вад. Діти з spina bifida (спинно-мозкова кила/відкритий мозок) деколи мають різновид клишоногості. Це спричинено пошкодженням спинальних нервів, що в свою чергу уражає долішні кінцівки. В іншому випадку, нормально зформовані стопи можуть стати покрученими через хворобу м’язів чи нервову хворобу.
Як клишоногість чи інші вади стопи лікуються?
Лікування клишоногості (еквіноварусної деформації стопи) розпочинається зразу після народження, часто вже на першому тижні життя. Його метою є поступово і лагідно поставити скручену стопу в позицію, з якої вона зможе рухатись вгору і вниз. За звичайним методом лікар повертає стопу вперед до тих пір, поки не відчувається біль, і фіксує її в такому положенні гіпсовою пов’язкою. Кожного тижня, спочатку, пов’язка змінюється таким чином, щоб наблизити стопу до її нормального (фізіологічного) положення. Після того, як стопу розігнули, її продовжують нахиляти далі вперед, щоб розтягнути закоротку зв’язку п’яти. Її утримують в цій позиції гіперкорекції декілька тижнів, щоб впевнитись, що стопа не повернеться у зворотній стан, після того як гіпс буде знято. Таке лікування звичайно триває від трьох до шести місяців. Потім на різний термін часу воно змінюється на ортопедичні скоби чи шини. З-за того, що клишоногість має схильність відновлюватись протягом перших сьоми років життя, дитина ще багато років потребує на медичний контроль.
Лікування клишоногості за допомогою гіпсових пов’язок буває корисним в декількох випадках. Однак, частіше зв’язка п’яти та інші зв’язки та м’язи литки та стопи є надто короткими, щоб розтягнути їх за допомогою гіпсування, і лікар має оперувати, щоб подовшти зв’язку. Хірургічне втручання також рекомендується у тих випадках, коли гіпсування не допомогає протягом перших трьох або шісти місяців життя.
Більшість немовлят з еквіноварусною деформаціїю стопи та аддукцією плесни не потребують лікування. Деколи батьки навчаються легким вправам на розтяжку, щоб прискорити видуження. Переважна більшість випадків еквіноварусної деформації стопи також проходить без лікування до трьох років. Однак, якщо стопа твердо утримується в ненормальній позиції, тобто у лікаря виникають складнощі під час корекції стопи у нормальне положення, рекомендується лікування. Це можуть бути фізичні вправи, корегуюче взуття чи гіпсування. У разі як стопа лишається негнучкою, лікування має бути розпочате до настання дитині восьми місяців, це дає кращі результати. Взагалі для аддукції плесни оперативне втручання не є необхідним.
Завдяки кваліфікованого раннього лікування більшість дітей, навіть які мали тяжкі ступені клишоногісті, зростають у звичайних черевиках, займаються спортом, живуть повноцінним активним життям. Проте, уражена стопа і нога в цілому не наздогоняють неуражену ногу за зростанням. Різниця звичайно незначна – 2,5см чи меньше, і розмір взуття відрізняється на один чи два номери. Нелікована, тяжка ступінь клишоногості лишається покрученою і кінцівка зростає у такому вигляді.
Чи можна запобігти клишоногості?
Незважаючи на те, що інвалідності як наслідка клишоногості найчастіше можна запобігти завдяки ранньому лікуванню, на сьогодні немає метода запобігання виникненню клишоногості. Генетичне консультування допоможе батькам зрозуміти важливі деталі щодо кожної вагітності, що призвели до появи клишоногості у дитини, а також перспективи лікування. Взагалі, якщо дитина має ізольовану клишоногість (інші вади розвитку відсутні), ризик її повторення у інших вагітностях є низьким (десь 3%).
Які наукові дослідження проводяться стосовно клишоногості?
Дослідження щодо виникнення і запобігання клишоногості зрідка спрямовані на окрему ваду. Вони звичайно охоплюють наукові розробки вроджених вад м’язів, нервів та кісток. М’язи та кістки ненародженої дитини можуть ушкоджуватись такою великою кількістю різних чинників, що потрібно буде ще декілька додаткових років досліджень, щоб винайти той чинник, що викликає клишоногість.
Переглянуто редакційною колегією I.B.I.S.: 17/10/2000
Дефект міжшлуночкової перетинки
(Ventricular Septal Defect)
Н.Г. Тандура
Лікар-неонатолог відділення недоношених дітей
Волинського обласного дитячого територіального медичного об’єднання
Визначення:
Дефект міжшлуночкової перетинки (ДМШП) – це вроджена вада серця (ВВС), при якій є патологічне сполучення між шлуночками серця, зі скидом крові зліва направо і збагаченим легеневим кровотоком.
Частота:
ДМШП серед ВВС зустрічається часто, 15-20%. На 10000 новонароджених припадає 20 випадків ДМШП.
Етіологія:
Численні етіологічні фактори розподіляються наступним чином: Хромосомна патологія – 5%. Серед синдромів аутосомної трисомії ДМШП найбільш часта причина серцевої аномалії. ДМШП виявляли у 47% пацієнтів з трисомією 13-15, в 74% з трисомією 18 і в 25% з трисомією 21. Мутація одного гену становить 2-3%. Фактори середовища (алкоголізм, краснуха, лікарські речовини та ін.) 1-2%.
Полігенно-мультифакторіальне успадкування становить 90%. Якщо в сім’ї є дитина з ВВС, ймовірність народження другої дитини з серцевою аномалією становить 2-5%. При наявності у двох родичів першого ступеня (батьки, брати, сестри) ВВС, ризик народження другої дитини з ВВС складає 15%. Мак-Мейон (Мас-Маhоn) рахує, що грає роль вік жінки, старші – мають більше шансів народити дитину з ВВС, ніж жінки молодого віку.
Патологія:
Міжшлуночкові дефекти найчастіше розміщуються в мембранозній частині перетинки – 70%.
Розмір дефекту від кількох міліметрів до 2 см в діаметрі. Приблизно 1/2 дефектів мають розмір 6-10 мм в діаметрі, четверта частина – менше 6 мм в діаметрі і четверта частина – більше 10 мм в діаметрі.
В результаті переваги тиску в великому колі кровообігу над малим відбувається скид крові зліва направо. Гіперволемія веде до легеневої гіпертензії, якщо діаметр дефекта більше 1/3-1/2 діаметра аорти. Гіпертензія може привести до набряку легень, якщо гідростатичний тиск в капілярах перевищує онко/осмотичний тиск крові. Ця загроза компенсується розвитком органічних змін в резистентних судинах (артеріолах). В міру вирівнювання тиску по обидві сторони перетинки серця скид крові зменшується. Коли легенево – судинний тиск стає вищим ніж системний, наступає реверсія шунта – венозна кров примішується до артеріальної. Ця заключна фаза динаміки легеневої гіпертензії одержала назву синдрома Ейзенменгера. Таким чином легенева гіпертензія проходить 3 фази: гіперволемічну, змішану (гіперволемія поєднується з підвищенням легенево-судинного тиску) і склеротичну (характеризується незворотніми змінами).
Клініка і діагностика:
Вважається, що у 2/3 пацієнтів ДМШП має безсимптомний перебіг.
Цю ВВС діагностують при аускультації серця – вислуховується грубий систолічний шум в 3-4 міжребер’ї зліва. Шум проводиться на спину.
ДМШП – це ВВС блідого типу, ціаноз відсутній. Фізичний розвиток дітей не страждає окрім випадків, коли дефект міжшлункової перетинки має великі розміри.
При ДМШП великих розмірів, крім систолічного шуму в 3-4 міжребер’ї, зліва від грудини вислуховується акцент 2 тону над легеневою артерією. Серце збільшене в розмірі. У дітей грудного і раннього віку відмічається задуха, виражена пітливість (результат гіперсимпатикотонії). Ці діти схильні до рецидивуючих пневмоній і інших захворювань органів дихання. Рано розвивається тотальна серцева недостатність: збільшення печінки, селезінки, ядуха, тахікардія, набряки, застійні вологі хрипи в легенях.
В більш старшому віці основні клінічні прояви вади зберігаються, але діти скаржаться на болі в серці, серцебиття, відстають у фізичному розвитку.
Між клінічними даними і даними ЕКГ існує кореляція. При невеликому дефекті ЕКГ нормальна. Ознаки ізольованої гіпертрофії міокарда лівого шлуночка, як правило, поєднуються з відносно великим дефектом, помірним скидом крові зліва направо без збільшення загальнолегеневого опору. Ознаки гіпертрофії міокарду правого шлуночка завжди супроводять легеневу гіпертензію. Клінічні симптоми комбінованої гіпертрофії міокарду обох шлуночків поєднуються з середнім та великим дефектом перетинки і легеневою гіпертензією різної важкості. На рентгенограмі органів грудної клітки при значному скиді крові легеневий малюнок посилений. Форма серця неспецифічна, талія згладжена, шлуночки збільшені, може бути вибухання дуги легеневої артерії, інколи аневризматичне.
На ехокардіографії, якщо дефект великий, при зміні кута локації від верхівки до основи серця може бути відсутнім ехосигнал від перетинки на деякому протязі. Діагноз встановлюють по збільшенню індекса правого шлуночка, збільшенню швидкості діастолічного руху мітрального клапана, збільшенню кінцево-діастолічного діаметра лівого шлуночка.
Катетеризацію порожнини серця і ангіографію проводили хворим з ДМШП при серцевій недостатності і/ або ознаках легеневої гіпертензії.
По даним I. Hoffman спонтанне закриття ДМШП буває в 45% випадків, невеликі дефекти міжшлункової перетинки закриваються, як правило, до 5-6 років.
Ускладнення:
Розвиток синдрому Ейзенменгера пов’язаний з розвитком склеротичної незворотної фази легеневої гіпертензії. В результаті високого тиску в правому шлуночку виникає скид крові справа наліво. У дитини з’являється ціаноз спочатку малинового кольору (ознака помірного недоокислення артеріальної крові киснем), потім синій і фіолетовий.
Клінічно зменшується шум, серцева недостатність, межі серця, що помилково розцінюється як покращення. В дійсності хворий стає іноперабельним. На ЕКГ при синдромі Ейзенменгера визначається гіпертрофія правих відділів серця, на рентгенограмі – збіднення легеневого малюнку на периферії при його посилені в прикореневій зоні, симптом “обрубаного дерева”, різке вибухання дуги легеневої артерії.
Може бути перехід ДМШП в “бліду форму” тетради Фалло. В зв’язку з турбулентністю потоку крові при скиді через дефект виникає підклапанний стеноз легеневої артерії, в результаті чого формується “набута” форма тетради Фалло. Клінічна картина вади змінюється: з’являється ціаноз, зменшуються розміри серця і ознаки серцевої недостатності, приєднується другий шум (шум стенозу легеневої артерії) в 3-му міжребер’ї зліва, який з часом стає домінуючим. На рентгенограмі змінюється конфігурація серця – воно набуває форми “чобітка”.
Слід також пам’ятати про можливість розвитку недостатності аортальних клапанів в 2-7% випадків ДМШП або приєднання бактеріального ендокардиту (частіше при невеликих дефектах).
Співвідношення статі: Ч1 : Ж1.
Генне картування і групи зчеплення генів: невідомі.
Лікування:
Терапевтичні заходи при ДМШП направлені на обмеження фізичного навантаження, стабілізацію гемодинаміки серцевими глікозидами і діуретиками, попередження і лікування супутніх захворювань.
Показами до оперативної корекції ДМШП у дітей перших років життя є легенева гіпертензія, рефрактерна недостатність серця, рецидивуючі пневмонії, гіпотрофія ІІ-ІІІ ст.
Література:
- Белоконь Л.А., Подзолков В.П. Врожденные пороки сердца.- Москва: Медицина,1991.- 351 с.
- Castaneda A, Jonas R, Mayer J, et al. Cardiac Surgery of the Neonate and Infante.- W.B. Sounders Co.,1994:506 p.
- Leavroudis C, Backer C. Pediatric Cardiac Surgery. Second Edition – MOSBY – Year Book, Inc.,1994:645 p.
Переглянуто редакційною колегією I.B.I.S.: 19/12/2001
Синдром Дауна – показники розвитку
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”.
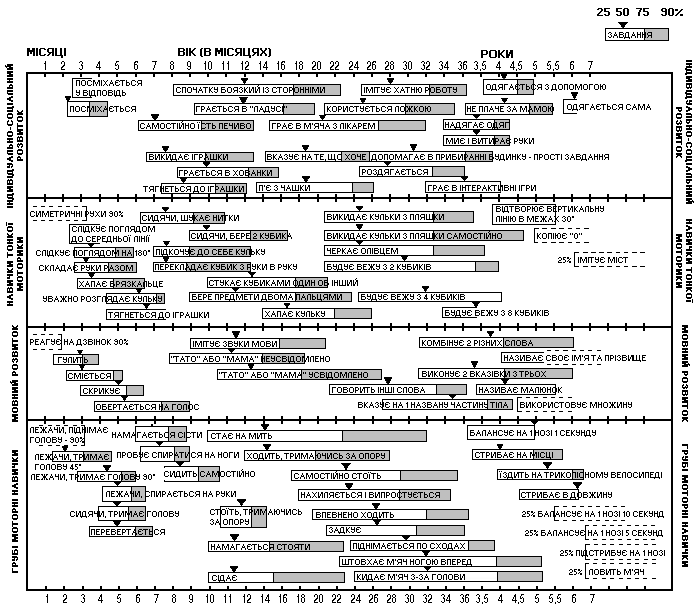
Таблиця оцінювання розвитку дітей з синдромом Дауна, які не виховувались в спеціальних установах. Дана таблиця була складена після обстеження 96 дітей з синдромом Дауна (57 хлопчиків та 39 дівчат), які проживали в м. Детройті і виховувались батьками вдома. Chen H та Wooley Jr. RV: Growth 42:157, 1978.
Синдром Дауна – довжина вуха
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”.
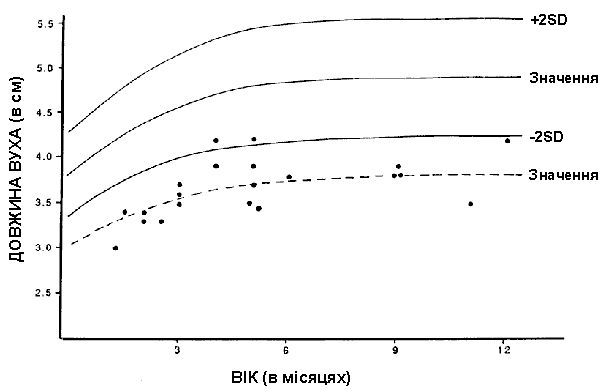
Довжина вуха у дітей з синдромом Дауна (виділено рисками та крапками) порівнюється з нормальним розвитком вух. Дані базуються на вимірюванні довжини вуха у 25 новонароджених та 28 дітей з синдром Дауна. Дані про довжину вуха у здорових дітей отримані в результаті обстеження 200 новонароджених з повним терміном гестації та 100 дітей віком до 12 років і більше. Aase JM et al: J Pediatr 82:845, 1973.
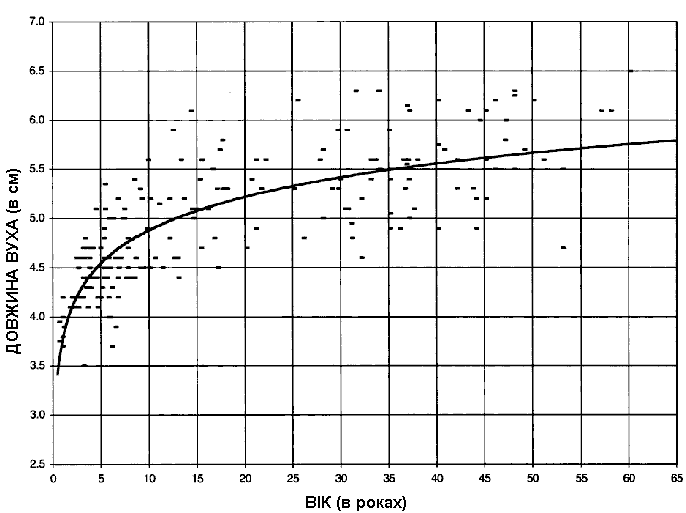
Вимірювання довжини вуха проводилось у 199 пацієнтів з синдромом Дауна. Дані зібрані Judith Allanson, MD, (1998).
|
|
|
|
|
|






{kind=link}