
Serhiy
Вимірювання тулуба і нижніх кінцівок
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”
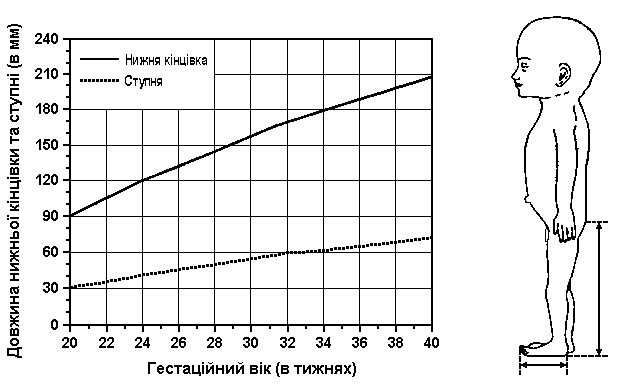

За Scammon та Calkins: The Development and Growth of the External Dimensions of the Human Body in the Fetal Period, University of Minnesota, 1929.
Кісти хоріоїдного сплетіння
(Choroid Plexus Cyst)
З.О. Сосинюк
Лiкар-генетик обласної медико-генетичної консультацiї
Рiвненського клiнiчного лiкувально-дiагностичного центру iменi Вiктора Полiщука
Включені:
- Кісти судинного сплетіння.
Виключені:
- Трисомія 18;
- Трисомія 21.
Частота:
Складає від 2 до 3,5% випадків при обстеженні плодів у другому триместрі вагітності.
Етіопатогенез:
Нейроепітелій судинних сплетінь по мірі свого росту утворює епітеліальні складки та вирости, які видаються всередину церебральних бокових вентрикул, створюючи так званий “хоріоїдальний ворс”, який в свою чергу перетворюється на канальці різних розмірів. Частина з них формує порожнини, обмежені сполучною тканиною і заповнені спинномозковою рідиною та продуктами розпаду клітин, які акумулюються і збільшують розміри кіст, потоншуючи власну стінку. Таким чином, кістами судинних сплетінь вважають утвори, діаметр яких перевищує розміри канальців у 5 разів.
Основні діагностичні критерії:
В тканині хоріоїдного сплетіння, частіше на рівні ділянок входу в бокові шлуночки, візуалізуються округлі ділянки з чіткими рівними контурами, зниженої ехощільності.
Кісти судинного сплетіння можна діагностувати з 12 тижнів вагітності, але оптимальний термін їх візуалізації – від 14 до 24 тижнів вагітності. Наприкінці першого триместру кісти можуть займати практично весь об”єм сплетіння. Із збільшенням терміну вагітності розміри судинних сплетінь зменшуються в порівнянні із розмірами шлуночків. Тому в другій половині другого триместру , коли розміри бокових шлуночків і судинних сплетінь невеликі, кісти судинного сплетіння візуалізувати складно. У більшості випадків діаметр кіст судинних сплетінь не перевищує 10мм, і вони спонтанно зникають до 26 тижнів вагітності. Вони можуть бути поодинокими, множинними, односторонніми, двосторонніми.
Клінічні ознаки:
Наявність малих за розміром кіст у тканині хоріоїдного сплетіння – часте явище при аутопсії (зустрічається у 50% випадків в усіх вікових групах населення). Таким кістам не надається клінічного значення тому, що більшість з них мають асимптомний перебіг. Однак, бувають випадки, коли виникає необхідність хірургічного втручання через порушення циркуляції спинномозкової рідини з розвитком обструктивної гідроцефалії.
Знахідки:
Згідно даним літератури, кісти судинного сплетіння діагностуються у 8% плодів з хромосомними аномаліями, при поєднаних аномаліях – в 46% випадків. Так, кісти судинного сплетіння є потенційними маркерами трисомії 18 і мають місце майже в 30% плодів з даною патологією. Тому при оцінці ризику трисомії 18 у зв’язку з наявністю у плодів кіст хоріоїдного сплетіння враховують, як вік матері, так і супутні аномалії. Значно рідше кісти хоріоїдного сплетіння зустрічаються при трисомії по 21 хромосомі.
Схема 1. Алгоритм обстеження вагітної при діагностуванні кіст хоріоїдного сплетіння у плода
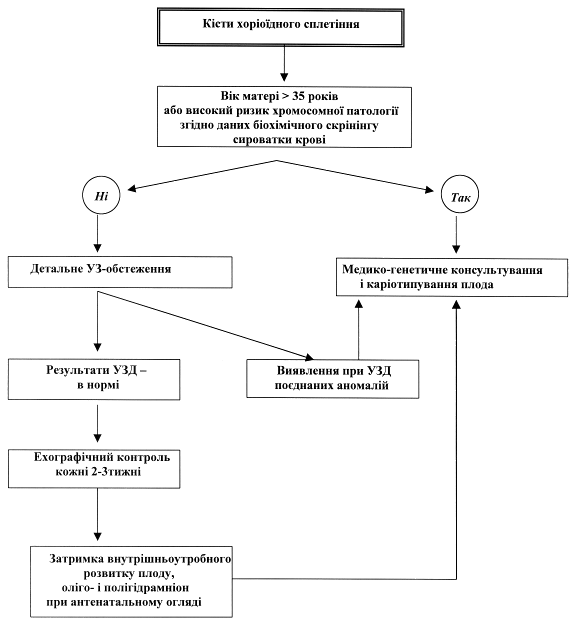
Пренатальна тактика:
При виявленні кіст хоріоїдного сплетіння необхідно провести детальну ультразвукову оцінку стану анатомії плоду і визначити рівень альфа-протеїну в крові вагітної. У випадку наявності поєднаних аномалій і відхиленні рівня материнського альфа-протеїну показане каріотипування плода. Якщо ж при сонографії не виявлено ніяких інших аномалій і результат біохімічного маркеру в нормі, слід проводити динамічний ехографічний контроль за станом ізольованих кіст хоріоїдного сплетіння кожні 2-3 тижні. Діагностика затримки внутрішньоутробного розвитку плоду чи багатовіддя є показом до амніоцентезу.
Акушерська тактика:
Кісти хоріоїдного сплетіння відносять до доброякісної патології. Показане динамічне ультразвукове обстеження за станом плоду і виключення гідроцефалії. Акушерська тактика в даному випадку стандартна.
Профілактика:
Рекомендоване медико-генетичне консультування та заходи преконцепції.
Список літератури:
- Janesh K Gupta, James G. Thornton, Richard J Lilford. Management of fetal choroid plexus cysts. British Journal of Obstetrics and Gynecology 1997;104:881-886.
- Janesh K Gupta, Mairead Cave, Richard J Lilford, Thomas A Farrell, Henry C Irving, Gerald Mason, Cathryn M Hau. Clinical significance of fetal choroid plexus cysts. Lancet 1995;346:724-729.
- Ronald C. Reinsch. Choroid plexus cysts – Association with trisomy: Prospective review of 16,059 patients. Obstetrics and Gynecology 1997;176:1381-3.
- Shawn Choong, Simon Meagher. Management of Choroid Plexus Cysts in the Mid-Trimester Fetus. Obstetrics and Gynecology 1999;39:1-7.
- Zerres K, Schuler H, Gembruch U, Bald R, Hansmann M, Schwanitz G. Chromosomal findings in fetuses with prenatally diagnosed cysts of the choroid plexus. Human Genetics 1992;89:301-304.
Переглянуто редакційною колегією I.B.I.S.: 15/02/2002
Вимірювання середнього пальця
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”

3, 50 та 97 перцентилі довжини середнього пальця відповідно до гестаційного віку. Довжина середнього пальця визначається відстанню між проксимальною згинальною складкою середнього пальця та його вільним краєм. Дані обстеження 87 доношених (48 хлопчиків та 39 дівчаток) та 111 недоношених (55 хлопчиків та 56 дівчаток) новонароджених 27-41 тижнів гестації у перші 36-60 годин життя. Жодна дитина не мала вроджених вад. Merlob P et al: Birth Defects: Orig Art Series XX(7):38, 1984.
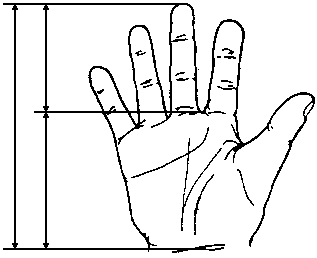

3, 50 та 97 перцентилі індексу довжини середнього пальця відповідно до гестаційного віку.
Індекс довжини середнього пальця =
Довжина середнього пальця
Довжина кисті
Див. пояснення вище. Merlob P et al: Birth Defects: Orig Art Series XX(7):38, 1984.
Синдром Кернса-Сейра
(Kearns-Sayre Syndrome)
Т.Б. Вінокурова
Лікар-генетик
Кримського республіканського спеціалізованого медико-генетичного центру
Синоніми:
Офтальмоплегія, пігментна дегенерація сітківки й кардіоміопатія; очно-черепно-соматичний синдром.
Основні діагностичні критерії:
- Вік хворого при маніфестації хвороби – 4-18 років.
- Прогресуюча зовнішня офтальмоплегія.
- Пігментний ретиніт + мінімум одна з наступних ознак:
- атаксія;
- інтенційний тремор;
- блокада серця;
- підвищення рівня білка у спинно-мозковій рідині більш, ніж 1 г/л;
- “рвані” червоні волокна в біоптатах м’язової тканини.
Клінічні прояви:
Клініка характеризується багатосистемністю уражень.
М’язова система:
- птоз, як правило, симетричний і білатеральний;
- повільно прогресуюча зовнішня офтальмоплегія;
- низхідний характер м’язової слабкості: обличчя-плече-тулуб-нижні кінцівки;
- зміна тембру голосу, поперхування, міалгії.
Нервова система:
- мозочкова атаксія;
- інтенційний тремор;
- розумова відсталість (в 50% випадків);
- спонтанні міоклонії;
- ураження периферичної нервової системи (поліневропатія, нейросенсорна туговухість у 2/3 хворих).
Орган зору:
- пігментний ретиніт;
- на очному дні пігментна грануляція типу “перець з сіллю”;
- атрофія диску зорового нерва (ДЗН);
- зниження гостроти зору.
Серцево-судинна система:
- синкопальний стан;
- брадикардія;
- дилятаційна кардіоміопатія;
- на ЕКГ – ураження провідної системи серця, зміна сегменту ST, ознаки дисфункції синусово-передсердного вузла.
Ендокринна система:
- низькорослість;
- гіпогонадизм;
- гінекомастія;
- цукровий діабет;
- гіперальдостеронізм;
- гіпопаратиреоз.
Факультативні ознаки:
- скелетні аномалії (кіфосколіоз, краніосиностоз, метафізарна дисплазія, гіпоплазія зубної емалі);
- ураження нирок (нирково-тубулярний ацидоз, синдром де Тоні-Дебре-Фанконі).
Існує повний і неповний синдром Кернса-Сейра. Повний синдром включає:
- хронічну прогресуючу зовнішню офтальмоплегію;
- пігментний ретиніт;
- A-V блокаду.
Дебют до 10 років, прогноз несприятливий. Неповна форма синдрому:
- Перший варіант:
- хронічна прогресуюча зовнішня офтальмоплегія;
- міопатичний симптомокомплекс по низхідному типу + один з облігатних симптомів;
- прогноз залежить від віку дебюту (більш несприятливий при пізньому дебюті) і ступеня залучення до патологічного процесу серцево-судинної системи.
- Другий варіант:
- ізольована хронічна зовнішня офтальмоплегія;
- Дебют пізній.
Диференціальний діагноз:
- Окуло-фарінго-дистальна міопатія, А/Д.
- Офтальмоплегія сімейна, статична, А/Д.
- Прогресуюча офтальмоплегічна м’язова дистрофія з ураженням черепних нервів і вадами спинних м’язів, А/Д.
- Офтальмоплегія прогресуюча сімейна із зниженням інтелекту й неврологічними симптомами, А/Д.
Етіологія:
Синдром пов’язаний з наявністю делеції мітохондріального геному, що частіше зачіпає гени 1 і 4 комплексів дихального ланцюга. У деяких хворих, окрім делеції, виявлена дуплікація мітохондріальної ДНК.
Важлива генетична особливість синдрому – співіснування нормальної і мутантної мітохондріальної ДНК.
Тип успадкування: переважна більшість випадків – спорадичні.
Співвідношення статей: Ч:Ж = 1:1.
Прогноз: несприятливий.
Лікування:
- дієта із зниженим вмістом вуглеводів;
- підвищення продуктивності електронно-транспортного ланцюга в тканинах організму та зниження вторинного дефіциту біологічно активних речовин:
- коензим Q10 (убіхинон) в дозі до 120 мг/добу (2 мг/кг) 3-6 міс.
- тіамін 900 мг/добу.
- α-карнітін 100 мг/добу.
- фолієва кислота 1 мг/добу.
- аскорбінова кислота 2-4 г/добу.
- вітамін Е до 300-500 мг/добу.
Виключаються лікарські препарати, здатні погіршити енергетичний метаболізм – хлорамфенікол, барбітурати, у разі оперативних втручань з обережністю призначаються анестезуючі речовини, які можуть негативно впливати на атріо-вентрикулярну провідність.
Номер з каталогу МІМ:
530000 Kearns-Sayre Syndrome, KSS.
Література:
- Вельтищев Ю.Е. Наследственная патология человека.– М., 1994.
- Николаева О.А., Темин П.А. Синдром Кернса-Сейра: генетические аспекты, клиника, возможности лечения //Рос. вестник перинатологии и педиатрии.- 1997.- № 2.
- Темин П.А., Казанцева Л.З. Наследственные нарушения нервно-психического развития детей.- М.: Медицина, 2001.
- Kearns-Sayre Syndrome (http://www.emedicine.com/ped/topic2763.htm).
- Kearns-Sayre Syndrome (https://www.omim.org/entry/530000).
Переглянуто редакційною колегією I.B.I.S.: 08/09/2007
Вимірювання кистей та нігтів пальців рук
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”
3, 50 та 97 перцентилі довжини кисті відповідно до гестаційного віку. Довжина кисті визначається відстанню між дистальною складкою зап’ястя та кінчиком середнього пальця. Дані обстеження 87 доношених (48 хлопчиків та 39 дівчаток) та 111 недоношених (55 хлопчиків та 56 дівчаток) новонароджених 27-41 тижнів гестації у перші 36-60 годин життя. Жодна дитина не мала вроджених вад. Merlob P et al: Birth Defects: Orig Art Series XX(7): 38, 1984.
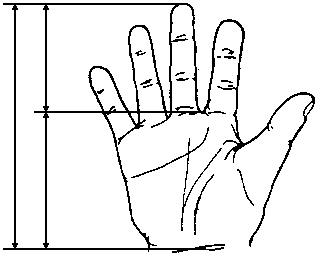

Виміри нігтів пальців рук і великого пальця
у доношених новонароджених
1 стандартне відхилення (мм) |
|
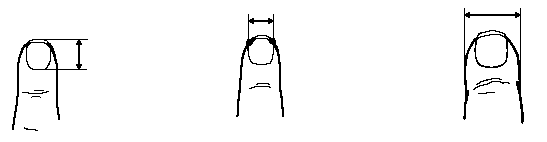
Вимірювання проводились у 120 здорових дітей 38-42 тижнів гестації у перші 3 дні життя. 52% оглянутих – дівчатка, 67% – темношкірих дітей.
Показник довжини нігтя визначався відстанню між серединою внутрішнього та зовнішнього краю нігтя. Показник ширини нігтя вимірювався у найширшому місці нігтя. Ширина великого пальця визначалась на рівні проксимального кінця нігтя. Подаються результати триразових вимірювань із використнням штангенциркуля. За Hudson VK et al: Dismorph Clin Genet 1:145, 1988
Синдром каудальної регресії
(Caudal Regression Syndrome)
О.Ф. Буга
Лікар-генетик
Шепетівської центральної районної лікарні Хмельницької області
Синоніми:
- каудальна дисплазія;
- сакральна агенезія;
- фокомелічна діабетична ембріопатія.
Основні діагностичні критерії:
- відсутність крижової кістки;
- відсутність поперекових хребців на різних рівнях;
- поєднані аномалії інших органів та систем.
Клінічна картина:
Клінічні прояви каудальної регресії залежать від рівня та величини дефекту. В залежності від важкості ураження клінічні прояви коливаються від нетримання сечі та калу до повної втрати чутливості та паралічу нижче рівня дефекту. Результатом відсутності крижової кістки є вирівнювання сідниць, вкорочення міжсідничної складки. При відсутності хребців поперекового відділу хребта клінічно спостерігається вкорочення тулуба. Як правило, атрофія нижніх кінцівок, згинально-ротаційно-відвідні контрактури кульшових, колінних суглобів. У важких випадках характерна так звана “поза Будди”.
Етіологія:
Невідома. Синдром каудальної регресії має виражену кореляцію з цукровим діабетом. Діабет вагітних, судинна недостатність є провокуючими факторами в розвитку каудальної регресії. Народження дітей з каудальною регресією пов’язано з діабетом, або предіабетом у матерів, що документально підтверджено різними дослідженнями. Цей синдром зустрічається у 200 разів частіше у дітей, народжених від матерів, що страждали цукровим діабетом.
Патогенез:
Вважається, що синдром каудальної регресії є наслідком дефекту мезодерми, який виникає на 4-ому тижні ембріонального розвитку і проявляється зміщенням зачатків нижніх кінцівок, що призводить до множинних дефектів нижнього відділу хребта з поєднаною неврологічною симптоматикою.
Поєднані симптоми:
- Кісткова система:
- деформація стопи;
- вивих кульшового суглобу;
- деформація кісток тазу;
- кіфосколіоз;
- відсутність ребер;
- щілина піднебіння;
- синдактилія, полідактилія.
- Шлунково-кишкова система:
- атрезія анального отвору;
- пахова грижа;
- дефект передньої черевної стінки;
- неповний поворот кишківника.
- Сечо-статева система:
- гідронефроз;
- підковоподібна нирка;
- агенезія однієї нирки;
- ектопія сечоводів;
- міхурово-сечовивідний рефлюкс;
- зміщення зовнішніх геніталій;
- відсутність Мюлєрової протоки.
- Нервова система:
- менінгоцеле;
- spina bifida;
- дефекти нервових стовбурів.
- Нориці:
- трахео-езофагальна;
- ректо-вагінальна;
- ректо-уретральна.
- Змішані:
- вроджені вади серця;
- косоокість.
Співвідношення статі: Ч 2,7 : Ж 1.
Спадковість:
Хоча генетичний фактор розвитку каудальної регресії не виявлений, більшість авторів схиляються до думки, що існує генетична схильність до розвитку патології.
Популяційна частота:
Синдром каудальної регресії зустрічається з частотою 0,1-0,25 : 10000 нормальних пологів. Частота розвитку синдрому у плодів, матері яких хворіють на цукровий діабет збільшується у 200-250 разів, в порівнянні із здоровою популяцією.
Вік маніфестації: з народження.
Лікування:
Якщо патологія виявлена пренатально в ранній термін гестації, вагітність доцільніше перервати.
При народженні живої дитини показана хірургічна корекція у спеціалізованих центрах для корекції наявних дефектів.
Прогноз:
Залежить від ступеня важкості дефекту крижової кістки та супутніх аномалій органів та систем. Виживання залежить від урологічної патології, рівня дефекту кісток хребта. Важкі форми поєднуються з серцевою, нирковою, дихальною патологією, які впливають на рівень ранньої дитячої смертності.
Пренатальна діагностика:
Виявлення синдрому каудальної регресії за допомогою УЗД залежить від протяжності та важкості дефекту. Ступінь дефекту коливається від повної відсутності крижової кістки в поєднанні з аномаліями поперекових хребців та іншими поєднаними вадами (див. вище), до аномалії крижової кістки без супутніх дефектів.
Найбільш типовим є виявлення при УЗД відсутності кількох хребців, щитовидний вигляд з’єднань між кістками, близьке розміщення крил здухвинних кісток, зменшення відстані між головками стегнових кісток. Часто спостерігається обмеженість рухів нижніх кінцівок плода. В першому триместрі пренатальна діагностика утруднена через неповну осифікацію крижової кістки. Вкорочення верхівки крижової кістки та ненормальний вигляд жовчного міхура запропоновані як рання ультразвукова ознака.
Постнатальна діагностика:
Ґрунтується на виявленні поєднаних аномалій у сукупності з рентгенологічним виявленням патології нижніх відділів хребта.
Диференційна діагностика:
Диференційну діагностику необхідно проводити з:
- асоціацією VATER;
- сірінгомієлією;
- хромосоми 13q синдромом;
- хромосоми 18 трисомії синдромом;
- хондродисплазіями.
Основною діагностичною відмінністю між перерахованими синдромами та синдромом каудальної регресії є наявність патології крижової кістки при останньому.
Номер з каталогу МІМ:
- 182940 Neural Tube Defects
- 134780 Femoral-Facial Syndrome; FFS
- 131240 Endothelin 1; EDN 1
- 115470 Cat Eye Syndrome, CES
Література:
- Adra et al. Caudal Regression Syndrome: Etiopathogenesis, PrenatalDiagnosis, and Perinatal Management. Obstetrical and Gynecological Survey 1994; Volume 49, Number 7.
- Goto et al. Diabetic Embriopathy. Current Science 1994;6:486-491.
- Nievelstein et al. MR of Caudal Regression Syndrome: Embriologic Implications. American Society of Neuroradiology June 1994;2.
- Welch et al. The Syndrome of Caudal Regression: A Review, Including Etiologic Considerations and Evidence of Heterogenety. Pediatric Pathology 1984;2:313-327.
Переглянуто редакційною колегією I.B.I.S.: 08/07/2002
Вимірювання передпліччя
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”

3, 50 та 97 перцентилі довжини передпліччя відповідно до гестаційного віку. Довжина передпліччя визначається відстанню між ліктьовим та шиловидним відростком променевої кістки, зігнутої під кутом 90 градусів руки. Дані обстеження 87 доношених (48 хлопчиків та 39 дівчаток) та 111 недоношених (55 хлопчиків та 56 дівчаток) ізраїльських новонароджених 27-41 тижнів гестації у перші 36-60 годин життя. Жодна дитина не мала вроджених вад. Merlob P et al: Birth Defects: Orig Art Series XX(7):37, 1984.


3, 50 та 97 перцентилі індексу довжини передпліччя відповідно до гестаційного віку.
Індекс довжини передпліччя =
Довжина передпліччя
Довжина верхньої кінцівки
Див. пояснення вище. Merlob P et al: Birth Defects: Orig Art Series XX(7):37, 1984.
Синдром Торіелло-Карі
(Toriello-Carey Syndrome)
Наталія Олексіївна Тищенко
Кафедра медичної генетики
Київської медичної академії післядипломної освіти імені П.Л. Шупика
Синдром Торіелло-Карі – це синдром множинних вад розвитку, маніфестація якого включає агенезію мозолистого тіла, незвичні риси обличчя, аномалад П’єра-Робена та інші аномалії.
Частота: описано близько 40 випадків.
Етіологія та патогенез: не з’ясовані.
Основні діагностичні критерії:
- Характерні риси обличчя: телекант, гіпертелоризм, короткі очні щілини, короткі та рідкі вії, маленький ніс, повні щоки, тонкі губи, опущені донизу кутики рота, щілина піднебіння, мікрогнатія, малі аномалії вушних мушель.
- Коротка шия.
- Постнатальна затримка зросту.
- Гіпотонія.
- Агенезія мозолистого тіла.
- Вроджені вади серця.
- Малі аномалії геніталій у хлопчиків.
- Малі аномалії кінцівок.
- Затримка розвитку.
Клініка:
При народженні більшість пацієнтів мають нормальні антропометричні параметри. У перші дні життя можуть спостерігатися респіраторний дистресс синдром, порушення ковтання, судоми. В більшості випадків розвивається постнатальна затримка фізичного розвитку, мікроцефалія, розумова відсталість. Характерною ознакою є великий розмір та пізнє закриття великого тім’ячка.
Аномалії очей включають гіпертелоризм, телекант, короткі очні щілини, птоз, короткі та рідкі вії. Загалом вроджені вади очних яблук не є характерними для синдрому Торіелло-Карі. Серед інших аномалій обличчя характерними є короткий ніс, щілина твердого або м’якого піднебіння, аномалад П’єра-Робена, повні щоки, тонкі губи, опущені донизу кутики рота, диспластичні вушні мушлі.
Вади розвитку нервової системи включають агенезію мозолистого тіла, гіпоплазію або атрофію мозочка, аномалію Денді-Уокера, колпоцефалію. Характерними є гіпотонія, зниження слуху, в деяких випадках можуть спостерігатися судоми або безсимптомні зміни на ЕЕГ. Найбільш типовими аномаліями гортані та глотки є гіпоплазія гортані, трахеомаляція, стридор, гіпотонія м’язів гортані, стеноз трахеї. Вроджені вади серця спостерігались у 33 з 45 описаних пацієнтів, переважно це дефекти перетинок, також стеноз легеневої артерії, відкрита артеріальна протока, тетрада Фалло, дисплазія клапанів, коарктація аорти.
Аномалії травної системи представлені дистопією анусу, пілоростенозом, хворобою Гіршпрунга. Характерною для синдрому ознакою є кили: пахова кила, пупкова кила, кила стравохідного отвору діафрагми. Більшість хлопців із синдромом Торіелло-Карі мають аномалії урогенітальної системи: крипторхізм, гіпоплазію геніталій (у дівчат гіпоплазія статевих губ).
Вади розвитку нирок нетипові, але описані гіпоплазія нирок та тазове розташування нирок.
Скелетні аномалії: коротка шия, порушення кількості ребер, вузька грудна клітка, аномалії ключиць, кіфосколіоз, порушення форми нижніх кінцівок. Також характерними є брахідактилія, клинодактилія, камптодактилія, тонкі пальці, синдактилія, широка відстань між І та ІІ пальцями стопи.
До інших ознак синдрому відносяться омфалоцеле, гіпоплазія тимуса, діафрагмальна кила, екзема.
Диференційний діагноз:
- Хромосомна патологія, включаючи теломерні делеції.
- Синдром Мардена-Уокера (MIM 248700).
- Синдром Ритчера-Шинцеля (MIM 220210).
- Синдром Паскуаля-Кастровійє.
Тип успадкування: аутосомно-рецесивний.
Вік маніфестації:
З народження. Пренатально діагноз можна запідозрити при виявленні агенезії мозолистого тіла та вроджених вад серця плоду при УЗД.
Лікування:
Симптоматичне. Хірургічна корекція вроджених вад розвитку. Розвиваючі психо-педагогічні програми.
Прогноз:
Залежить від спектру вроджених вад розвитку, які присутні в кожному випадку. Найстарший пацієнт був описаний в віці 14 років.
Профілактика: не розроблена.
Номер з каталогу МІМ:
217980 Corpus Callosum, Agenesis of, with Facial Anomalies and Robin Sequence.
Література:
- Gorlin RG, Cohen MM, Jr, Hennekam RCM. Syndromes of the Head and Neck. Fourth Edition. Oxford University Press. 2001:924-925.
- Toriello HV, Carey JC et al. Toriello-Carey syndrome: Delineation and review. Am J Med Genet 2003;123A:84-90.
- Wegner KJ, Hersh JA. Toriello-Carey syndrome: An additional case and summary of previously reported cases. Clin Dysmorph 2001;10:145-148.
Переглянуто редакційною колегією I.B.I.S.: 17/01/2006
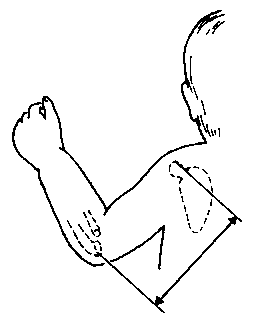
Вимірювання плеча
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”
3, 50 та 97 перцентилі довжини плеча відповідно до гестаційного віку. Довжина плеча визначається відстанню між акроміоном та ліктьовим відростком зігнутої під 90 градусів руки. Дані обстеження 87 доношених (48 хлопчиків та 39 дівчаток) та 111 недоношених (55 хлопчиків та 56 дівчаток) новонароджених 27-41 тижнів гестації у перші 36-60 годин життя. Жодна дитина не мала вроджених вад. Merlob P et al: Birth Defects: Orig Art Series XX(7):36, 1984.

3, 50 та 97 перцентилі індексу довжини плеча відповідно до гестаційного віку.
Індекс довжини плеча =
Довжина плеча
Довжина верхньої кінцівки
Див. пояснення вище. Merlob P et al: Birth Defects: Orig Art Series XX(7):36, 1984.

Кардіо-фаціо-кутанеальний синдром
(Cardiofaciocutaneous Syndrome)
О.П. Семененко
Лікар-генетик
Черкаського обласного перинатального центру
Синоніми:
- Cardiofaciocutaneous syndrome;
- CFC syndrome.
Синдром вперше описаний у 1986 р.
Частота:
Остаточна невідома (Reynolds J.F. та співавтори). Описано близько 35 випадків.
Основні діагностичні критерії:
- Вроджена вада серця.
- Ектодермальная дисплазія.
- Виступаюче чоло.
Клінічна характеристика:
- Зріст:
- Постнатальна затримка росту.
- Голова та обличчя:
- Відносна макроцефалія з великим виступаючим чолом.
- Звуження черепа у скроневих ділянках.
- Гіпертелоризм.
- Птоз.
- Екзофтальм.
- Короткий ніс з відкритими до переду ніздрями.
- Виступаючий фільтр.
- Серце:
- Дефект міжпередсердної перетинки.
- Стеноз легеневої артерії.
- Шкіра та волосся:
- Рідке кучеряве волосся.
- Повільний ріст волосся.
- Відсутність вій та брів.
- Атопічний дерматит.
- Гіперкератоз.
- Іхтіоз.
- Нервова система:
Неврологічна симптоматика пов’язана з аномаліями розвитку головного мозку: атрофією кори, гіпоплазією лобної долі, атрофією стовбура й аномаліями шлуночкової системи мозку. Загальним і характерним проявом є мовні порушення. Затримка інтелектуального розвитку, гіпотонія, ністагм, страбізм, гідроцефалія.
Поєднані симптоми:
Мікроцефалія, судоми, аномалії ЕЕГ, гіпертонія, глухота, підслизиста щілина піднебіння, кили, спленомегалія, кавернозна гемангіома, дисплазія нігтів.
Лікування:
- Симптоматичне.
- Хірургічна корекція вроджених вад розвитку.
- Розвиваючі психо-педагогічні програми.
Профілактика: не розроблена.
Етіологія:
Більшість випадків спорадичні. Ген локалізований на 7q34.
Номер з каталогу МІМ:
115150 Cardiofaciocutaneous syndrome.
Список літератури:
- Jones KL. Smith’s Recognizable Patterns of Human Malformation. 5th ed. Philadelphia: W. B. Saunders Company 1997:126-127.
- Reynolds JF, et al. New multiple congenital anomalies/mental retardation syndrome with cardio-cutaneous involvement – the CFC syndrome. Am J Med Genet 1986 Nov;25(3):413-27.
Переглянуто редакційною колегією I.B.I.S.: 12/12/2007
|
|
|
|
|
|





