
Serhiy
Синдром мікрофтальмії Ленца
(Lenz Microphthalmia Syndrome)
Л.Ф. Мавлюдова
Лікар-генетик
м. Євпаторія (Республіка Крим)
Синоніми:
- Анофтальмія з асоційованими аномаліями.
- Дисплазія Ленца.
Основні критерії:
Синдром мікрофтальмії Ленца (LMS) характеризується одно- чи двобічною мікрофтальмією та/чи анофтальмією у поєднанні з вродженими вадами вух, зубів, пальців, хребта, сечостатевої системи.
Асоційовані вади:
Аномалії очей. Мікрофтальмія часто супроводжується мікрокорнеа і глаукомою. Приблизно у 60% пацієнтів зустрічається колобома райдужної оболонки, іноді анофтальмія. Може спостерігатися колобома ціліарного тіла, судинної оболонки та диска зорового нерва. [Ng та ін. 2002]
При зниженні зору спостерігається ністагм. Нормальний чи зменшений розмір очного яблука може викликати вторинне зменшення очних щілин і зростання країв повік (анкілоблефарон).
Аномалії вух. Низькорозташовані, диспластичні вушні раковини, ротовані вперед, часом спостерігається туговухість.
Аномалії зубів. Агенезія верхніх різців, дефекти нижніх різців (широкі міжзубні проміжки).
Аномалії кістково-м’язової системи. Довга циліндрична грудна клітка з похилими, вузькими плечима, кифосколіозом, вираженим поперековим лордозом. Подвоєння великих пальців рук, вкорочення окремих пальців рук і ніг. Низький зріст.
Сечостатеві аномалії включають гіпоспадію, двосторонній крипторхізм, ниркову гіпоплазію/аплазію і гідроуретер.
Нервова система. Затримка розумового розвитку різного ступеня вираженості.
Анатомічні зміни ЦНС: спостерігається відсутність чи гіпоплазія зорових нервів і оптичної хіазми, крім того гіпоплазія мозолистого тіла. Можливі також затримка моторного розвитку та порушення циркадних ритмів – чергування циклів сну та активності. При вираженій розумовій відсталості у хворих спостерігається порушення поведінки, схильність до самопошкодження.
Результати обстежень:
При “простій мікрофтальмії” чи “чистій мікрофтальмії” зменшене в розмірах очне яблуко має всі анатомічні елементи і зберігає деякий зір. Легка форма простої мікрофтальмії діагностується шляхом вимірювання осьової довжини очного яблука при ультразвуковому дослідженні. Осьова довжина складається із довжини переднього сегмента (від переднього краю корнеальної вершини до краю верхівки лінзи) і наступного сегмента (від краю піка лінзи до переднього краю сітківки). Скоректована вікова загальна довжина очного яблука менше п’ятого перцентиля свідчить на користь діагнозу мікрофтальмії [Weіss та ін. 1989]. Для дорослих загальна осьова довжина менше 18,5 мм підтверджує діагноз [Weіss та ін. 1989].
Ультразвукове обстеження нирок необхідно для виявлення ниркової аплазії, гіпоплазії та гідроуретера.
Етіологія:
Вважається, що два гени, локалізовані на X-хромосомі ANOP1 (Xq27-q28) і ANOP2 (Xp11.4-p21.2) можуть бути зв’язані з синдромом мікрофтальмії Ленца.
Тип успадкування: зчеплений з Х-хромосомою рецессивний.
Рекурентний ризик:
Ризик для сибсів залежить від того, чи є мати носієм, чи ні. Якщо мати є носієм, ризик передачі складає 50% при кожній вагітності. Особи чоловічої статі, що успадковують цю мутацію будуть хворими; сибси жіночої статі, що успадковують цю мутацію, будуть носіями. Більшість чоловіків із синдромом Ленца не мають потомства. Якщо мати не є носієм, ризик для сибсів – низький.
Молекулярно-генетичне обстеження для синдрому мікрофтальмії Ленца можливе у науково-дослідних установах.
Частота виникнення:
Поширення і розподіл серед етнічних груп невідоме. Зареєстровані випадки захворювання у європейців, жителів Африки, латиноамериканців [Hoefnagel та ін. 1963, Ogunye та ін., 1975, Brunquell та ін., 1984, Ng та ін., 2002]. Передбачувана частота вродженої мікрофтальмії серед немовлят в Европі – 1,2-1,8 на 10000.
Диференційний діагноз:
Мікрофтальмія, успадковувана Х-зчеплено рецесивно, вимагає диференціювання з аутосомно-рецесивною та аутосомно-домінантною формами. Це часом досить складно в сім’ях з обтяженим сімейним анамнезом. Мікрофтальмія може бути викликана хромосомними аномаліями. Хромосомні аномалії, пов’язані з мікрофтальмією, включають трисомії 13 і 18; 18p, 13q і 4p (включаючи синдром Wolf-Hіrschhorn); і тетрасомію 22q11.2. Ряд тератогенів, таких як спирт, талідомід, ізоретиноєва кислота, цукровий діабет у матері, перенесена під час вагітності краснуха. Повідомляється про амніотичні перетяжки, що спричинили виникнення мікрофтальмії [Warburg 1993].
Номер з каталогу МІМ:
309800 Microphthalmia Syndromic 1; MCOPS1.
Література:
- Brunquell PJ, Papale JH, Horton JC, Wіllіams RS, Zgrabіk MJ, Albert DM, Hedley-Whyte ET. Sex-lіnked heredіtary bіlateral anophthalmos. Pathologіc and radіologіc correlatіon. Arch Ophthalmol. 1984;102:108-13.
- Forrester S, Kovach MJ, Reynolds NM, Urban R, Kіmonіs V. Manіfestatіons іn four males wіth and an oblіgate carrіer of the Lenz mіcrophthalmіa syndrome. Am J Med Genet. 2001;98:92-100.
- Graham CA, Redmond RM, Nevіn NC. X-lіnked clіnіcal anophthalmos. Localіzatіon of the gene to Xq27-Xq28. Ophthalmіc Paedіatr Genet. 1991;12:43-8.
- Hoefnagel D, Keenan ME, Allen FH. Heredofamіlіal bіlateral anophthalmіa. Arch Ophthalmol. 1963;69:760-764.
- Lehman DM, Sponsel WE, Stratton RF, Mensah J, Macdonald JC, Johnson-Paіs TL, Coon H, Reveles XT, Cody JD, Leach RJ. Genetіc mappіng of a novel X-lіnked recessіve colobomatous mіcrophthalmіa. Am J Med Genet. 2001,101:114-9.
- Lenz W. Recessіv-gesclechtsgebundene mіkrophthalmіe mіt multіplen mіsbіldungen. Z Kіnderheіlkd. 1955;77:384-390.
- Ng D, Hadley DW, Tіfft CJ, Bіesecker LG. Genetіc heterogeneіty of syndromіc X-lіnked recessіve mіcrophthalmіa-anophthalmіa: Іs Lenz mіcrophthalmіa a sіngle dіsorder? Am J Med Genet. 2002;110:308-14.
- Ogunye OO, Murray RF, Osgood T. Lіnkage studіes іn Lenz mіcrophthalmіa. Hum Hered. 1975;25:493-500.
- Warburg M. Classіfіcatіon of mіcrophthalmos and coloboma. J Med Genet. 1993;30:664-9.
- Weіss AH, Kousseff BG, Ross EA, Longbottom J. Sіmple mіcrophthalmos. Arch Ophthalmol. 1989;107:1625-30.
Переглянуто редакційною колегією I.B.I.S.: 19/03/2004
Тім’ячка, черепні шви та краніосиностоз
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”
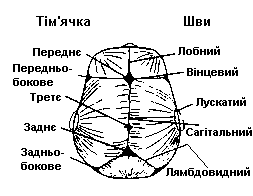
Зображення кісток, відкритих швів та тім’ячок черепу здорового новонародженого (вигляд зверху).
|
Переднє тім’ячко
Заднє тім’ячко Передньо-бокове тім’ячко Задньо-бокове тім’ячко Лобний шов Клінічне закриття швів Анатомічне закриття швів |
1 рік ± 4 місяці
Народження ± 2 місяці До 3 місяця Протягом 2 року життя До 3 року (у 10% ніколи) 6-12 місяців До 30 року |
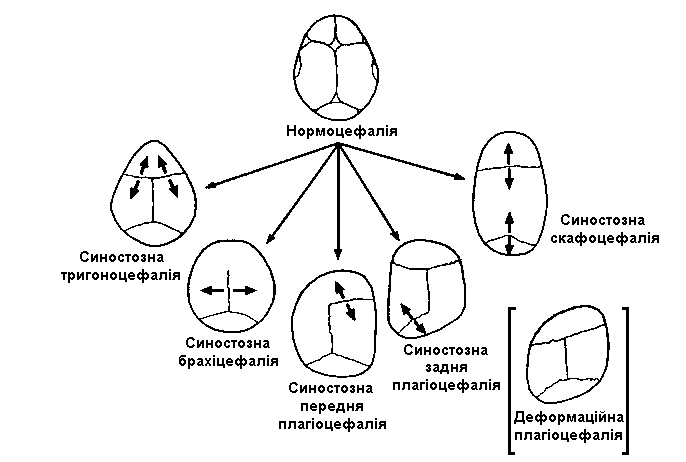
При синостозній скафоцефалії сагітальний шов передчасно закривається. У випадку синостозної брахіцефалії та синостозної передньої плагіоцефалії вінцевий шов закривається з одного чи з обох боків. При синостозній трігоноцефалії передчасно закривається лобний шов. В рідкісних випадках задньої синостозної плагіоцефалії лямбдовидний шов закривається однобічно. Такий стан часто помилково плутають з деформаційною плагіоцефалією (в квадратних дужках), при якій усі шви відкриті. Ступінь вираженості деформації черепу залежить від часу синостозування. Воно може бути вродженим або розвинутись в період новонародженості. Більше того, при залученні до процесу синостозування двох і більше швів, кінцева форма черепа може бути змінена. У підписах до малюнків використовуються прикметник синостозний, оскільки брахіцефалія, доліхоцефалія та трігоноцефалія можуть виникати і без синостозу.
Ілюстрація у верхньому лівому кутку за Cohen MM: Craniosynostosis: Diagnosis, Evaluation, and Management, Raven Press, 1986, p 39. Ілюстрація знизу за Cohen MM Jr: Anatomic, Genetic, Nosologic, and Psychosocial Considerations. В книзі: Craniosynostosis: Diagnosis, Evaluation, and Management, M.M. Cohen Jr and R.E. Maclean, Editors, Second Edition, Oxford University Press, New York, 1999, Ch. 11. Copyright © 1999 by Oxford University Press, Inc. Використано з дозволу Oxford University Press, Inc.
Метахроматична лейкодистрофія
(Metachromatic Leukodystrophy)
Н.О. Пічкур
Лікар-невролог
кандидат медичних наук
Н.В. Ольхович
Завідуюча медико-генетичною лабораторією
медико-генетичного центру УДСБ “ОХМАТДИТ”, м.Київ
Включення: метахроматична лейкодистрофія.
Основні критерії діагностики:
Прогресуюча втрата психо-мовних та рухових навичок, деменція, аномальна поведінка, полінейропатія, зниження активності цереброзидсульфатази, деструктивні зміни в білій речовині головного мозку.
Клінічні особливості:
Метахроматична лейкодистрофія об’єднує сім захворювань, які класифіковані залежно від віку початку хвороби та біохімічного дефекту. Найбільш часті форми асоціюються з дефіцитом арилсульфатази А і розподіляються на вроджену, пізню інфантильну, ранню ювенільну, пізню ювенільну та дорослу форми.
Метахроматичне забарвлення нервової тканини вперше було описано Альцгеймером у 1910 році у дорослого пацієнта з клінічними ознаками генералізованого паралічу. В 1921 році Вітте описав аналогічний клінічний випадок та виявив у хворого накопичення метахроматичного матеріалу не тільки в тканинах мозку, а й в нирках, печінці та тестикулах. У 1925 році Шольц детально описав трьох дітей з однієї родини, які мали прогресуючу лейкодистрофію, і припустив, що мієлінова аномалія, яка спостерігалась у цих дітей, повинна приводити до ушкодження гліальних клітин. Пайфер та Хірш у 1955 році, використовуючи заморожені препарати оригінальних пацієнтів Шольца, відкрили явище метахромазії. Тип МЛД, що починається в пізньо-інфантильному періоді був описаний Грінфілдом у 1933році, вроджений тип МЛД був описаний Файгіним у 1954 році. Яцкевич та Аустин проаналізувавши та дослідивши всі відомі на той час випадки довели, що так звану “метахроматичну лейкоенцефалопатію” слід класифікувати як ліпідоз. У 1963 році Аустин повідомив про дефіцит арилсульфатази А, який призводить до накопичення кислих ліпідів у тканинах пацієнтів та обумовлює ефект метахромазії.
Пізніше був знайдений термостабільний фактор, який активує арилсульфатазу А, і його дефіцит обумовлює картину так званого псевдодефіциту, який був діагностований у незначної кількості пацієнтів з МЛД. Інший рідкісний варіант хвороби, який асоціювався з ознаками мукополісахаридозів, був описаний у 1965 році і відомий як множинна сульфатазна недостатність. Біохімічну основу цього типу обумовлює дефіцит щонайменше семи різних сульфатаз.
Вроджена форма МЛД:
Виділення вродженої форми МЛД базується на гістологічному дослідженні двох випадків. Прямий біохімічний доказ накопичення сульфатидів в тканинах або ферментативного дефекту відсутній. Метахроматичний матеріал був знайдений скрізь в білій речовині, додатково були виявлені збільшені нейрони та визначався гліоз в корі головного мозку. Клінічні прояви характеризувалися нападами апноє, ціанозом та тоніко-клонічними судорогами, тонус м’язів був знижений. Обоє дітей померли у віці 6 тижнів. Можливо ці випадки були ранніми клінічними проявами пізньо-інфантильної форми і тому вроджена форма МЛД вважається багатьма авторами лише припущенням.
Пізньо-інфантильна форма:
Починається між першим і другим роками життя. У деяких пацієнтів затримка психо-моторного розвитку відмічається вже до року. У цих дітей уповільнюється розвиток мовлення, пізніше формуються навички самостійної ходи. Спостерігаються часті падіння та хода навшпиньки. В деяких випадках хода не формується взагалі. Симптоми захворювання можуть провокуватися інфекційними захворюваннями з наступним відновленням функцій, але з поверненням симптомів через декілька тижнів і значним прогресом симптомів хвороби.
В незначній кількості випадків хвороба розвивається як периферійна нейропатія. Клінічний перебіг захворювання можна поділити на 4 стадії.
Перша стадія. Розпочинається у віці 2-3 роки: дитина втрачає здатність самостійно сідати та утримувати позу при сидінні, іноді розвивається епілептичний синдром. Сухожилкові рефлекси на ранніх стадіях захворювання дещо знижені, м’язовий тонус має тенденцію до гіпотонії, спостерігається розвиток комплексу патологічних ступневих знаків. Перша стадія триває від кількох місяців до року.
Друга клінічна стадія. Характеризується значним розумовим регресом. Виникає дизартрія та афазія. Спостерігаються дегенеративні зміни на очному дні: зміна рожевого забарвлення дисків зорового нерву на сірий колір, з’являється ністагм. Тонус м’язів в ногах підвищується, можуть виникати інтермітуючі болі в кінцівках, що обумовлено ураженням периферійних нервів. Прогрес симптомів швидкий і займає не більше кількох місяців.
Третя клінічна стадія. Характеризується розвитком тетраплегії, псевдобульбарними та бульбарними порушеннями, що призводить до ускладнень при прийомі їжі. Прояви деменції значно виражені, але дитина здатна посміхатися і впізнавати батьків.
Фінальна 4-а стадія. Позначається втратою будь-якого контакту з хворим. Вони сліпі, не розуміють зверненої мови і не можуть розмовляти та рухатись. Годування таких пацієнтів проводиться за допомогою гастрального або назогастрального зонду. Тривалість цієї стадії від кількох місяців до кількох років.
Ювенільна форма МЛД:
Дебют ювенільної форми МЛД припадає на вік від 4 до 12 років. Розвиток хвороби у ранньому шкільному віці перш за все характеризується плутаниною у виконанні доручень, появою денного тривалого сну. Поряд з аномальними формами поведінки, рухи дитини стають незграбними, мова нерозбірлива. В неврологічному стані з’являються симптоми екстрапірамідної недостатності: зміна м’язового тонусу та тремор. Через рік дитина не може самостійно ходити та обслуговувати себе, спостерігаються судоми. Захворювання при ювенільній формі прогресує повільніше. Описані випадки тривалості хвороби до 20 років. Загалом тривалість життя у таких пацієнтів не перевищує 20 років.
Доросла форма МЛД:
Доросла форма МЛД може початись у будь-якому віці після пубертатного періоду. Описані випадки початку захворювання як у 15-16 років, так і в 62 роки. Початок захворювання супроводжується змінами особистості, розвитком депресивних станів, погіршенням пам’яті, зниженням темпів мислення. У пацієнтів можуть з’явитися симптоми деперсоналізації або паранойї. Втрачаються навички охайності. Неврологічний стан характеризується проявами пірамідної недостатності, ознаками невропатії. У фінальних стадіях захворювання розвивається тетрапарез, генералізовані епілептичні судоми, атрофія зорових нервів. Тривалість хвороби становить від 5 до 10 років.
Етіологія:
Розвиток метахроматичної лейкодистрофії обумовлений мутаціями в гені арилсульфатази А(ARSA). Ген ARSA (MIM ID: 250100) локалізований на 22 хромосомі людини в локусі 22q13.3, має довжину 3,2 тис.п.н. та містить 8 екзонів. На 1.05.2002 року описана 91 мутація в гені ARSA, що визначає розвиток метахроматичної лейкодистрофії. Серед них – 71 однонуклеотидна заміна (місенс- та нонсенс-мутації), 8 сплайсінгових мутацій, 10 делецій, із них 6 зі зсувом рамки зчитування, 1 інсерція та 1 інверсія. Більшість мутацій в гені ARSA знаходять у поодиноких випадках МЛД або в групах споріднених індивідуумів, що скоріше за все, є результатом природного мутаційного процесу. Найбільш розповсюдженими, тобто мажорними є сплайсингова мутація в 2-му інтроні (IVS2+1G) та місенс мутація в 8-му екзоні (P426L) гену ARSA.
МЛД зумовлена гомозиготністю одного аутосомно-рецесивного гену.
Частота виникнення: 1:40000.
Співвідношення статей: Ч1:Ж1.
Лікування:
Лікування симптоматичне, яке включає знеболюючі, протисудомні та транквілізатори. У літературі є повідомлення про проведення у таких хворих трансплантації кісткового мозку від матері пробанду. Спостерігалось уповільнення прогресу захворювання строком на 1 рік.
Диференційний діагноз:
Глобоідно-клітинна лейкодистрофія, лейкоенцефаліт Шільдера, інші види лейкодистрофій, шизофренія.
Номер за каталогом МІМ:
250100 Metachromatic Leucodystrophy.
Nt |
a.a. |
МУТАЦІЇ |
|||||
|---|---|---|---|---|---|---|---|
Література:
- Barth ML, Fensom A, Harris A. Prevalence of common mutations in the arylsulfatase A gene in metachromatic leukodystrophy patients diagnosed in Britain. Hum Genet 1993;91(1):73-77.
- Baxevanis A.D. The molecular biology database collection: 2002 update. Nucl Acid Res 2002;30(1):1-12.
- Berger J, Leoschl B, Bernheimer H, Lugowska A, Tylki-Szymanska A, Gieselmann V, Molzer B. Occurrence, distribution, and phenotype of arylsulfatase A mutations in patients with metachromatic leukodystrophy. Am J Med Genet 1997;69(3):335-340.
- Coulter-Mackie MB, Gagnier L, Beis MJ, Applegarth DA, Cole DE, Gordon K, Ludman MD. DNA-based diagnosis of arylsulfatase A deficiencies as a supplement to ensyme assay: a case in point. Clin Biochem 1997;30(1):57-61.
- Gieselmann V, Polten A, Kreysing J, von Figura K. Arylsulfatase A pseudodeficiency: loss of a polyadenylation signal and n-glycosylation site. Proc Nat Acad Sci USA 1989;86:9436-9440.
- Gort L, Coll MJ, Chabras A. Identification of 12 novel mutation and two new polymorphisms in the arylsulfatase A gene: haplotype and genotype-phenotype correlation studies in Spanish metachromatic leukodystrophy patients. Hum Mutat 1999;14(3):240-248.
- Hasegawa Y, Kawame H, Eto Y. Mutations in the arylsulfatase A gene of Japanese patients with metachromatic leukodystrophy. DNA Cell Biol 1993;12(6):493-498.
- Halsall DJ, Halligan EP, Elsey TS, Cox TM. Metachromatic leucodystrophy: a newly identified mutation in arilsulphatase A, D281Y, found as a compound heterozygote with I179L in adult onset case. Hum Mutat 1999;14(5):447.
- Holve S, Hu D, McCandless SE. Metachpomatic leukodystrophy in the Navajo: fallout of the American-Indian wars of the nineteenth century. Am J Med Genet 2001;101(3):203-208.
- Holve S, Hu D, McCandless SE. Metachpomatic leukodystrophy in the Navajo: fallout of the American-Indian wars of the nineteenth century. Am J Med Genet 2001;101(3):203-208.
- Kreysing J, von Figura K, Gieselmann V. Structure of the arylsulfatase A gene. Eur J Biochem 1990;191:627-631.
- Lissens W, Vervoort R, Van Regemorter N, Van Bogaert P, Freund M, Verellen-Dumoulin C, Seneca S, Liebaers I. A D255H substitution in the arylsulphatase A gene of two unrelated Belgian patients with late-infantile metachromatic leukodystrophy. J Inherit Metab Dis 1996;19(6):782-786.
- Polten A, Fluharty AL, Fluharty CB, Kappler J, von Figura K, Gieselmann V. Molecular basis of different forms of metachromatic leukodystrophy. N Engl J Med 1991;324:18-22.
- Regis S, Filocamo M, Stroppiano M, Corsolini F, Gatti R. Molecular analysis of the arylsulphatase A gene in late infantile metachromatic leukodystrophy patients and healthy subjects from Italy. J Med Genet 1996;33(3):251-252.
- Zlotogora J, Furman-Shaharabani Y, Harris A, Barth ML, von Figura K, Gieselmann V. A single origin for the most frequent mutation causing late infantile metachromatic leucodystrophy. J Med Genet 1994;31(9):672-674.
Переглянуто редакційною колегією I.B.I.S.: 20/12/2005
Синдром Ноя-Лаксової
(Neu-Laxova Syndrome)
Світлана Євгеніївна Оніщенко
Лікар-генетик
Херсонської обласної дитячої лікарні
У літературі цей синдром вперше описаний Річардом Л. Ноєм у 1971 році. Далі досліджено Ренатою Лаксовою та Вірою Повисіловою.
Визначення:
Cиндром Ноя-Лаксової – це рідкісний летальний синдром, що характеризується іхтіозом, набряком шкіри, дисплазією обличчя, внутрішньоутробною затримкою росту, мікроцефалією, аномаліями центральної нервової системи, гіпоплазією або ателектазами легень, контрактурами кінцівок, полігідрамніоном і короткою пуповиною.
Синоніми:
- Синдром Ноя
- Синдром Ноя–Повисілової
- Мікроцефалія, затримка росту, флексорні деформації
Популяційна частота:
Популяційна частота даного синдрому невідома, він вважається рідкісним. Повідомлялось про підвищену частоту захворювання серед пакистанців.
Етіологія:
Аутосомно-рецесивне захворювання. Багато пацієнтів народжені у близькоспоріднених шлюбах.
Клінічні ознаки синдрому:
Синдрому притаманна гетерогенність проявів:
- У деяких випадках зустрічаються набряки підшкірної клітковини, у інших такі прояви відсутні.
- Повторні спонтанні аборти.
- Гіпоплазія плаценти.
- Коротка пуповина зустрічається у 35% випадків захворювання. У деяких випадках пуповина має тільки 2 судини. При доношеній вагітності вага новонародженого становить 850-1500 гр. Довжина тіла новонародженого – 25-30 см. Майже всі пацієнти народжуються мертвими, або помирають через декілька годин після народження. Найдовше дитина із синдромом Ноя-Лаксової прожила 8 тижнів.
Багато ознак, таких як полігідрамніон, внутрішньоутробна затримка росту, генералізований набряк, деформації та набряк кінцівок, остеопенія і коротка тривалість життя пояснюються втратою протеїну через тріщини шкіри. Деякі структурні деформації можуть бути наслідками внутрішньоутробної мобільності.
Лицьові дизморфії
| Мікроцефалія | |
| Скошене чоло | |
| Сплощений ніс з широким переніссям | |
| Низько розташовані вушні раковини | |
| Екзофтальм | |
| Гіпертелоризм | |
| Мікрогнатія | |
| Коротка шия |
Передчасне закриття швів та тім’ячок, можливо, внаслідок малого розміру мозку. Ніздрі зазвичай малі, або, зрідка, майже відсутні. Повні щоки. Верхні та нижні повіки вивернуті і дуже гіпопластичні, здається, ніби вони повністю відсутні. Вушні раковини дуже часто є великими та деформованими. Губи вивернуті. Спостерігається двостороння розщілина губи та піднебіння.
М’язово-скелетні деформації. У багатьох пацієнтів короткі контрактовані кінцівки. Набряк на кистях рук та стопах. Гіпопластичні пальці, з нігтями або без них у 60% пацієнтів. Часто пальці заходять один на один. У 50% пацієнтів – “ступні-качалки”. Характерні синдактилія пальців рук та ніг, флексорне розташування пальців. Вузький таз. Контрактури суглобів.
Центральна нервова система. Важка мікроцефалія (у 85%). Різноманітні варіанти аномалій, такі як атрофія мозку, агенезія мозолистого тіла, гіпоплазія мозочка (у 40%), розширення бокових шлуночків (у 25%), ліссенцефалія (у 40%), голопрозенцефалія, агенезія хробака мозку. Зменшена кількість ядер у клітинах моста, мозочка та основі мозку.
Сечостатеві ознаки. У близько 35% пацієнтів спостерігається як однобічна агенезія нирки, так і гіпоплазія зовнішніх статевих органів.
Шкіра. У 50% пацієнтів – іхтіоз, набряк підшкірної клітковини.
Аномалії серця. Зустрічається дефект міжшлуночкової перетинки, дефект міжпередсердної перетинки, відкрита боталова протока, транспозиція магістральних судин.
Інші симптоми. Щілина губи та піднебіння зустрічається у приблизно 25% пацієнтів.
Диференційна діагностика:
- Церебро-окуло-фаціальний синдром (COFS).
- Вроджений іхтіоз.
Пренатальна діагностика:
Можлива, починаючи з 6-8 тижнів вагітності, коли спостерігається затримка внутрішньоутробного розвитку плоду. На 12-16 тижнях – недостатня рухова активність кінцівок плоду, на 16-24 тижнях – фаціальні та скелетні аномалії, аномалії центральної нервової системи, нирок, серця та ін. З 34 тижня – фетальна едема, гіпертелоризм та екзофтальм.
Нижче наводимо власні спостереження синдрому Ноя-Лаксової. Пренатальна діагностика синдрома Ноя-Лаксової:




Фенотип пробанда із синдромом Ноя-Лаксової:

Прогноз: 100% летальність.
Номер з каталогу МІМ:
256520 Neu-Laxova Syndrome; NLS.
Література:
- Козлова С. И., Демикова Н.И, Семанова Е., Блинникова О.Е. Наследственные синдромы. – М.: Практика,1996.- С. 191-192.
- Мислицький В.Ф., Пішак В.П., Проняєв В.І. Спадкові синдроми. Епонімічний словник-довідник.– Чернівці: Прут,1998.– С. 1.
- Curry CJR. Further comments on the Neu-Laxova syndrome. (Letter) Am J Med Genet 1982;13:441-444. [Medline]
- Fitch N, Curry C. Comments on Dr. Curry’s classification of the Neu-Laxova syndrome. (Letter) Am J Med Genet 1983;15:515-518. [Medline]
- Fitch N, Resch L, Rochon L. The Neu-Laxova syndrome: comments on syndrome identification. Am J Med Genet 1982;13:445-452. [Medline]
- Kainer F, Prechtl HFR, Dudenhausen JW, Unger M. Qualitative analysis of fetal movement patterns in the Neu-Laxova syndrome. Prenatal Diag 1996;16:667-669. [Medline]
- Karimi-Nejad MH, Khajavi H, Gharavi MJ, Karimi-Nejad R. Neu-Laxova syndrome: report of a case and comments. Am J Med Genet 1987;28:17-23. [Medline]
- King JA, Gaedner V, Chen H, Blackburn W. Neu-Laxova syndrome: pathological evaluation of a fetus and review of the literature. Pediatr Pathol Lab Med 1995;15:57. [Medline]
- Kuseyri F, Bilge I, Bilgic L, Apak MY. Neu-Laxova syndrome: report of a case from Turkey. Clin Genet 1993;43:267-269. [Medline]
- Laxova R, Ohdra PT, Timothy JAD. A further example of a lethal autosomal recessive condition in sibs. J Ment Defic Res 1972;16:139-143. [Medline]
- Lazjuk GI, Lurie IW, Ostrowskaja TI, Cherstvoy ED, Kirillova IA, Nedzved MK, Usoev SS. The Neu-Laxova syndrome–a distinct entity. Am J Med Genet 1979;3:261-267. [Medline]
- Manning MA, Cunniff CM, Colby CE, El-Sayed YY, Hoyme HE. Neu-Laxova syndrome: detailed prenatal diagnostic and post-mortem findings and literature review. Am J Med Genet 2004;125A:240-249. [Medline]
- Meguid NA, Temtamy SA. Neu-Laxova syndrome in two Egyptian families. Am J Med Genet 1991;41:30-31. [Medline]
- Mueller RF, Winter RM, Naylor CPE. Neu-Laxova syndrome: two further case reports and comments on proposed subclassification. (Letter) Am J Med Genet 1983;16:645-649. [Medline]
- Muller LM, de Jong G, Mouton SCE, Greeff MJ, Kirby P, Hewlett R, Jordaan HF. A case of the Neu-Laxova syndrome: prenatal ultrasonographic monitoring in the third trimester and the histopathological findings. Am J Med Genet 1987;26:421-429. [Medline]
- Naveed, Manjunath CS, Vijaya S. New manifestations of Neu-Laxova syndrome. Am J Med Genet 1990;35:55-59. [Medline]
- Neu RL, Kajii T, Gardner LI, Nagyfy SF, King S. A lethal syndrome of microcephaly with multiple congenital anomalies in three siblings. Pediatrics 1971;47:610-612. [Medline]
- Ostrovskaya TI, Lazjuk GI. Cerebral abnormalities in the Neu-Laxova syndrome. Am J Med Genet 1988;30:747-756. [Medline]
- Povysilova V, Macek M, Salichova J, Seemanova E. Fatal syndrome of multiple malformations in 3 siblings. Cesk Pediat 1976;31:190-194. [Medline]
- Rouzbahani L. New manifestations in an infant with Neu Laxova syndrome. (Letter) Am J Med Genet 1995;56:239-240. [Medline]
- Scott CI, Louro JM, Laurence KM, Tolarova M, Hall JG, Reed S, Curry CJR. Comments on the Neu-Laxova syndrome and CAD complex. Am J Med Genet 1981;9:165-175. [Medline]
- Shapiro I, Borochowitz Z, Degani S, Dar H, Ibschitz I, Sharf M. Neu-Laxova syndrome: prenatal ultrasonographic diagnosis, clinical and pathological studies, and new manifestations. Am J Med Genet 1992;43:602-605. [Medline]
- Spranger JW, Schinzel A, Myers T, Ryan J, Giedion A, Opitz JM. Cerebroarthrodigital syndrome: a newly recognized formal genesis syndrome in three patients with apparent arthromyodysplasia and sacral agenesis, brain malformation and digital hypoplasia. Am J Med Genet 1980;5:13-24. [Medline]
- Winter RM, Donnai D, Crawfurd Md’A. Syndromes of microcephaly, microphthalmia, cataracts, and joint contractures. J Med Genet 1981;18:129-133. [Medline]
Переглянуто редакційною колегією I.B.I.S.: 20/12/2005
Крипторхізм
(Cryptorchidism)
Людмила Миколаїівна Зайцева
Лікар-неонатолог
Волинського обласного дитячого територіального медичного об’єднання
Вступ:
Хоча крипторхізм є одним із найбільш розповсюджених вад чоловічого сечостатевого тракту, його проблеми залишаються суперечливими та далекими від вирішення. Це проблеми пренатальної діагностики, діагностики взагалі, вибору консервативного лікування чи оперативної корекції, реабілітації, попередження онкозахворювання та порушень фертильності, можливість прогнозування цієї патології по спадковості, та зв’язок крипторхізму з великою групою генетичнодетермінованих станів.
Визначення:
Крипторхізм – це природжене системне захворювання, одним із проявів якого є неопущення яєчок. Яєчко закладається біля медіальної поверхні первинної нирки і потім, до 7 місяця внутрішньоутробного розвитку, переміщується до калитки. Вважається, що шлях яєчок визначається кількома механізмами, які включають внутрішньочеревний тиск, локальну наявність тестостерону, стан gubernaculums testis та його прикріплення до калитки, а також зміни придатка яєчка. Нормальний розвиток яєчок визначається їх локалізацією всередині калитки. Температура калитки на 2-3°С нижче, ніж у черевній порожнині. Це стає вирішальним для можливості нормального сперматогенезу. Розрізняють псевдокрипторхізм, справжній крипторхізм моно- або білатеральний та відсутність яєчок. У випадку справжнього крипторхізму яєчка розташовані повздовж нормального шляху їх опущення, але через вкорочення сім’яного канатика яєчко не досягло порожнини калитки і розташоване інтраабдомінально, в паховому каналі, або над калиткою. Псевдокрипторхізм включає випадки “мігруючого яєчка” (втягування яєчка в черевну порожнину при переохолодженні, збудженні), та ектопію яєчка (його розташування в стегновому каналі, в періанальній зоні, біля кореня статевого члена). Відсутність яєчок має місце у 20-40% випадків і зустрічається як однобічна, або двобічна аномалія.
Етимологія:
Грецька kryptos – таємний + orchis – яєчко.
Поширеність:
340:10000. Поширеність крипторхізму корелює з віком та вагою. Так у ваговій категорії 451-910 гр. крипторхізм зустрічається у 100% випадків, 911-1810 гр – 62%, 1811-2040 – 25%, 2041-2490 – 17%, 2491-2720 – 12%, 2721-3630 – 3,3%, 3631-5210 – 7%. У річному віці частота крипторхізму зустрічається у 0,7-0,8%, шкільному віці – 0,76-0,95%, у дорослих – 0,7-1%
Етіологія: невідома.
Патогенез:
Невизначений. Було запропоновано 3 теорії: відсутність або аномалія gubernaculums testis, вроджений гонадний дефект і дефіцит гормональної гонадотропної стимуляції.
Вроджений гонадний дефект. В цій теорії яєчка є нечутливі до гонадотропіну, чим можна пояснити виникнення монолатерального чи білатерального крипторхізму.
Дефіцит гонадотропної гормональної стимуляції. Материнський хоріальний гормон стимулює продукцію андрогенів в останні місяці гестації. При відсутності такого впливу має місце білатеральний крипторхізм. Ця теорія пояснює високу поширеність крипторхізму у передчасно народжених немовлят.
Асоційовані аномалії:
Крипторхізм зустрічається при 258 синдромах. Висока частота при:
- Аарського синдромі;
- Голопрозенцефалії;
- Коккейна синдромі;
- Дубовіча синдромі;
- Гіпопітуітаризмі;
- Кальмана синдромі;
- Мекккеля-Грубера синдромі;
- Нунан синдромі;
- Опіца синдромі;
- Синдромі Прадера-Віллі;
- Лоуренса-Муна-Бідла синдромі;
- Триплоідії;
- Трисомії по 13, 18 парі;
- 4р (Вольфа-Хіршхорна);
- 13q, 18q;
- гіпофізарній аплазії,гіпоплазії;
- тестикулярній ферментній недостатності;
Центральна нервова система. Порушення гіпоталамо-гіпофізарно-яєчникової стимуляції можуть асоціюватися із крипторхізмом. Вони включають таку патологію як: аненцефалія з недорозвитком гіпофіза, гіпофізарну аплазію і spina bifida, а також Кальмана синдром (гіпогонадотропізм, гіпогонадизм і аносмія).
Сечовий тракт. У 3-17% знаходять аномалії сечовивідного тракту, такі як мальротації, підковоподібну нирку, гіпоспадію та агенезію нирок. У більшості випадків плоди з пухлиною Вільмса мали білатеральний крипторхізм, порівнюючи із загальною чисельністю (278:10000).
Диференціальна діагностика:
З псевдокрипторхізмом або фізіологічним крипторхізмом.
Діагностика:
- Фізикальне обстеження;
- УЗД;
- Магнітно-резонансна томографія;
- Комп’ютерна томографія черевної порожнини і органів малої миски;
- Лапароскопія;
- Реноехографія;
- Екскреторна рентгенографія;
- Визначення рівня гіпофізарних гормонів(лютропіну, фолітропіну, тестостерону).
Прогноз:
- Безпліддя. Гістологічні зміни зі сторони сім’яників прямо пропорційні дистопії та відстані між яїчком та калиткою.
- Новоутворення. Крипторхізм в 35-48 разів підвищує ризик розвитку раку статевих клітин.
- Інші ускладнення. Перекрут яєчка, виникнення пахової кили.
Повторний ризик:
Ряд випадків крипторхізму успадковується по аутосомно-домінантному типу. Аналізуючи випадки крипторхізму було встановлено, що 1,5-4% батьків і 6% братів також мали цю патологію. В першу чергу обтяжена спадковість по чоловій лінії оцінюється як 0,67±0,16.
Лікування:
Лікування крипторхізму може бути терапевтичним (гормональним) або хірургічним. Оптимальним для лікування є вік від 6 до 18 місяців. На першому етапі показане терапевтичне лікування, оскільки тестостерон являється одним із стимуляторів нормального процесу опущення яєчок. Використовують хоріонічний гонадотропін. Успіх його використання обмежений із-за небажаних побічних ефектів: передчасне закриття зон росту та пришвидшення статевого дозрівання. Хірургічна корекція: орхіпексія та герніопластика. Однак, орхіпексія, створюючи сприятливі топографічні умови для сперматогенної та ендокринної функції яєчка, одночасно посилює зрушення в імунологічному гомеостазі, важливій ланці у механізмі розвитку крипторхізму, як аутоімунного, так аутоагресивного захворювання. Тому доцільне комплексне лікування із використанням препаратів пасивного посилення супресорної імунологічної активності, при виявлені гіпофункції яєчок через 3-6 місяців після орхіпексії призначають хоріоідний гонадотропін, стимулятори гормонопоезу та репаративних процесів у яєчку.
Номер з каталогу МІМ:
219050 Cryptorchidism, Unilateral or Bilateral.
Література:
- Ерохин А.П., Воложин С.И. Крипторхизм.- М., 1995.- С.344.
- Райцина С.С. Сперматогенез и структурные основы его регуляции.- Москва, 1985.- С. 206.
- Czeizel A, Erodi E, Toth J. Genetic of undescended testis. J Urol 1981;126:528-529.
- Rajfer J. Congenital Anomalies of the testis. In : Walsk DC, Retick AB,Saney TA. Cambell’s Urology.- W.B.Saunders, Harcourt.- 1992:1543-1562.
Переглянуто редакційною колегією I.B.I.S.: 27/08/2002
Зріст, вага та обвід голови у дорослих
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”
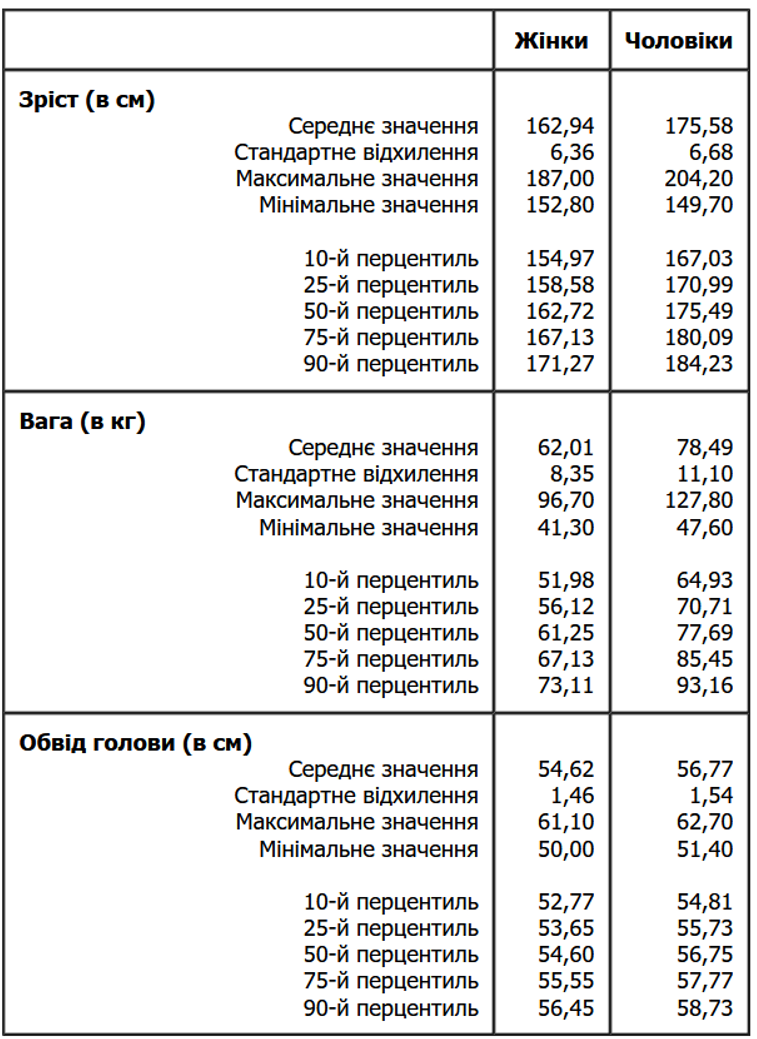
Результати антропометричного огляду військовослужбовців армії США у 1987-1988 роках (1774 чоловіків та 2208 жінок відповідного віку та етнічної належності); червень 1988 року. Gordon CC et al.: 1988 Anthropometric Survey of U.S. Army Personnel: Summary Interim Report (NAT- ICK/TR- 89/027), Technical Report, March 1989, pp. 107, 145, 170.
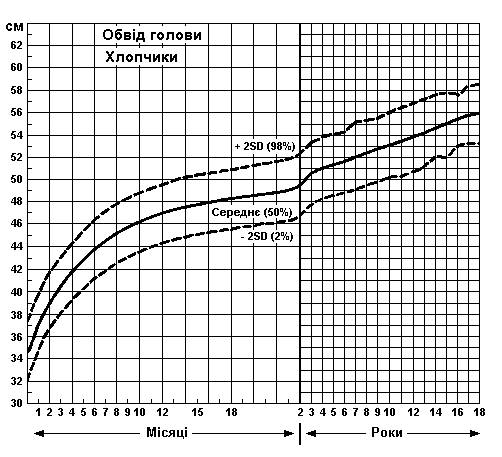
Обвід голови у хлопчиків та дівчат
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”

Синдром Фразера
(Fraser Syndrome)
О.П. Семененко
Завідувач
Черкаським обласним медико-генетичним центром
Синоніми:
- Криптофтальм з іншими мальформаціями.
- Cryptophthalmos with other malformations.
- Криптофтальму–синдактилії синдром.
- Cryptophthalmos–syndactyly syndrome.
Частота:
Популяційна частота невідома, вважається, що синдром зустрічається рідше ніж 1:100000 народжень. Повідомлялось, що частота синдрому у понад 100 разів вища у популяції циган. Описано близько 117 пацієнтів (А.М.Slavotinek і C.J.Tifft в 2002 р.).
Основні діагностичні критерії: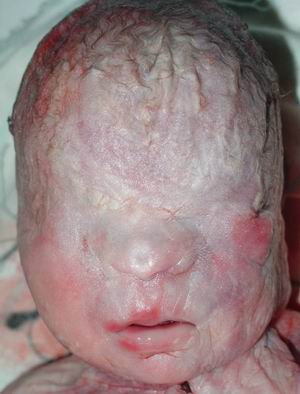
- Великі критерії:
- Криптофтальм (прикрите шкірою око, відсутність брів та вій, аномалії сльозової протоки): повний, неповний, прихований.
- Синдактилія (шкірна).
- Аномалії геніталій (маленький пеніс, гіпоспадія, крипторхізм, збільшення клітора, атрезія вагіни).
- Сібс з синдромом Фразера.
- Малі критерії:
- Аномалії вуха (дизморфічні вушні мушлі, кондуктивна туговухість).
- Аномалії носа (широке пласке перенісся, гіпоплазія ніздрів).
- Аномалії гортані (стеноз, атрезія).
- Щілина губи і/або піднебіння.
- Дефекти скальпа(незвичайний латеральний ріст волосся).
- Пупкова кила.
- Агенезія нирок ( або гіпоплазія).
- Розумова відсталість.
- Mікроцефалія, менінгомієлоцелє.
Для діагностики синдрому Фразера мінімальними діагностичними ознаками можуть бути два великі та один малий критерій, або один великий і чотири малі критерії.
Розвиток:
Прогноз щодо здоров’я та життя зумовлений важкістю вроджених вад, насамперед, гортані та нирок. Загалом, близько 25% дітей із синдромом Фразера гинуть антенатально. 20% відсотків дітей помирають до року (частіше від дефектів гортані), часто в перші тижні життя. Агенезія або двобічна дисплазія нирок мають несприятливий прогноз. Часто зустрічаються вроджені вади серця (гіпертрофічна кардіоміопатія, аномалія Ебштейна, коарктація аорти, дефект міжшлуночкової перетинки, єдиний артеріальний стовбур, транспозиція магістральних судин, найбільш частою вадою при синдромі Фразера є відкрита артеріальна протока). Прогноз більш сприятливий, якщо криптофтальм є єдиною вадою, але зір буде низьким навіть після хірургічної корекції. Однак затримка розвитку спостерігається й більшості пацієнтів, що вижили.
Лікування: хірургічна корекція.
Профілактика:
Медико-генетичне консультування, враховуючи аутосомно-рецесивний тип успадкування обмеження дітонародження, допоміжні репродуктивні технології (донація сперми), пренатальная діагностика плода. Пренатальна діагностика синдрому Фразера можлива з 18 тижнів вагітності через ультразвукову детекцію деяких ВВР або ж їх поєднання: мікрофтальм, синдактилія, обструктивна уропатія, легенева обструкція через атрезію гортані, асцит, водянка плоду, набряк шиї, олігогогідрамніон (внаслідок порушеного розвитку нирок).
Етіологія:
Аутосомно-рецесивний тип успадкування. Синдром обумовлений мутацією FRAS1 гена або FREM2 гена. Існують припущення, що цей ген обумовлює порушення процесу апоптозу (процесу запрограмованої смерті клітин) в уражених осіб. Якщо деякі клітини не проходять нормального процесу апоптозу в ембріональному періоді, то це зумовлює надлишковий ріст деяких тканин, як от повік при криптофтальмі чи міжпальцевих перетинок у випадку синдактилії.
Номер з каталогу МІМ:
219000 Fraser Syndrome.
Література:
- Белозеров Ю.М. Детская кардиология. Наследственные синдромы.- 2008.- С. 359-360.
- Boyd PA, Keeling JW, Lindenbaum RH. Fraser syndrome: a revier of eleven cases with postmortem findings. Am J Med Genet 1988;31:159-68.
- De Jong A, Warren M, Rehrauer W, Harter J, Baraboo M, Chandra S, Pauli RM, Singer DB, Fritsch MK. Fraser syndrome: affected siblings born to nonconsanguineous parents and diagnosed at autopsy. Pediatr Dev Pathol 2008 May-Jun;11(3):220-5.
- Herrera-Soto JA, Price CT. The presence of bilateral hip dysplasia and genu valgum in Fraser syndrome. Orthopedics 2008 Jan;31(1):81.
- Kiyozumi D, Takeichi M, Nakano I, Sato Y, Fukuda T, Sekiguchi K. Basement membrane assembly of the integrin α8β1 ligand nephronectin requires Fraser syndrome-associated proteins. J Cell Biol 2012 May 28;197(5):677-89.
- Slavotinek AM, Tifft CJ. Fraser syndrome and cryptophthalmos: review of the diagnostic criteria and evidence for phenotypic modules in complex malformation syndromes. J Med Genet 2002 Sep;39(9):623-33.
- Smith’s Recognizable Patterns of Human Malformation. 5th Edition. 1997:308-309.
- Van Haelst MM, Scambler PJ; Fraser Syndrome Collaboration Group, Hennekam RC. Fraser syndrome: a clinical study of 59 cases and evaluation of diagnostic criteria. Am J Med Genet A 2007 Dec 15;143A(24):3194-203.
Переглянуто редакційною колегією I.B.I.S.: 21/09/2012
Крижово-куприкова тератома
(Sacrococcygeal Teratoma)
Олена Андріївна Мельничук
Лікар-неонатолог
Волинської обласної дитячої клінічної лікарні
Визначення:
Тератома (від грецького “teratos”, що означає “монстр”) – ембріональна доброякісна пухлина, яка містить похідні трьох зародкових листків та звичайно виникає як утвір в крижово-куприковій ділянці плода.
Частота:
Частота крижово-куприкових тератом становить 1:35000 – 1:40000 новонароджених. У плодів жіночої статі вони зустрічаються частіше (до 80% випадків), але у плодів чоловічої статі більше виражена тенденція до злоякісного перебігу.
Етіологія:
Точна етіологія невідома, більшість дослідників вважають виникнення тератоми спорадичним.
Тип спадкування:
Невідомий. Декілька родин з аутосомно-домінантно-успадкованою пресакральною тератомою були описані в літературі, однак ризик повторного виникнення тератоми в даних випадках вірогідно не вищий за загальний фон.
Основні діагностичні критерії:
Підвищений рівень материнського альфа-фетопротеїну, полігідрамніон повинні бути обов’язковим показом до ретельного ультразвукового обстеження.
Клініка:
Локалізація крижово-куприкових тератом може бути різною: вони можуть бути переважно зовнішніми (47%), частково розташовуватись в тазовій ділянці (34%), в черевній та тазовій порожнинах (9%), пресакрально без зовнішних проявів (10%). Пухлина складається з щільних (кістки, волосся, зуби) та кистозних утворів. Поверхня покрита незміненою шкірою, в деяких випадках – шкіра витончена, виразково змінена, з гемангіоматозними ділянками.
Супутні вроджені аномалії зустрічаються у 15% пацієнтів, частіше це урогенітальні, аноректальні вади, спиномозкова кила.
Ускладнення:
- серцево-судинна недостатність внаслідок великого кровотоку в судинах тератоми при значних її розмірах (більше 10 см в діаметрі);
- масивний крововилив або кровотеча з пухлини, які можуть настати спонтанно внутрішньоутробно або бути викликаними родовою діяльністю матки;
- механічна травма пухлини, в тому числі розрив під час пологів;
- прогресуюча водянка плоду та плаценти, яка може призводити до внутрішньоутробної загибелі плоду;
- непрохідність кишківника та сечових шляхів внаслідок механічного зміщення та перетискання останніх пухлиною;
- септичні ускладнення при значних порушеннях цілісності пухлини;
- ризик малігнізації внаслідок морфологічної різноманітності та незрілості тканин, з яких складається тератома.
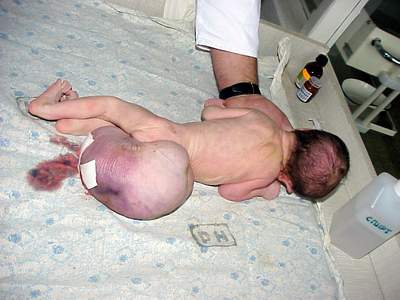 Крижово-куприкова тератома у дитини,
Крижово-куприкова тератома у дитини,
народженої у Волинській області
Диференційний діагноз необхідно проводити з:
- менінгомієлоцеле;
- ректальним абсцесом;
- дермоїдною кистою;
- ангіомою;
- ліпомою;
- неврогенною пухлиною.
Лікування:
Дитина з тератомою крижово-куприкової ділянки повинна бути прооперована в перші 2 тижні життя, а при наявності ускладнень – в перші дні. За даними Арканзаського медичного університету (1999 р.), хірургічне лікування крижово-куприкової тератоми повинно бути якнайскорішим. Якщо пухлину діагностовано внутрішньоутробно, рекомендується інтраматкове видалення як тератоми, так і всього куприка. Невидалення куприка призводить до 30-40% ризику рецидиву. Значні розміри тератоми, загроза розвитку серцево-судинної недостатності плоду є показами до ембріонального хірургічного втручання. Ця процедура розроблена та успішно застосовується в Каліфорнійському університеті. Поряд з цією методикою пропонується лікування за допомогою радіочастотного впливу: пункція матки через передню черевну стінку з посиланням радіочастотних хвиль через голку до пухлини, в результаті дії високої температури знищуються кровоносні судини, які живлять тератому, зупиняється її ріст. Протипоказом до ембріонального хірургічного втручання є водянка плоду, полігідрамніон, значне потовщення плаценти, а також материнська прееклампсія.
Прогноз:
Прогноз залежить від розмірів та гістології пухлини, ступеню її незрілості, супутніх вроджених вад, механізму родорозрішення та своєчасного повного хірургічного видалення. В 90% випадків хворі виживають. Невидалені протягом 2 місяців тератоми малігнізуються в 7-10%, протягом 1 року – в 37%, протягом 2 років – в 50%.
Профілактика: невідома
Спеціальні рекомендації:
Всі плоди з діагностованою тератомою повинні підлягати детальній сонографічній оцінці з метою підтверження діагнозу, виключення супутніх аномалій, оцінки розмірів плаценти, типу пухлини, наявності або відсутності водянки плоду. Виходячи з даних обстежень вибирається тактика ведення вагітності, родорозрішення, терміни та методики лікування.
Література:
- Баиров Г.А. Срочная хирургия детей: руководство для врачей.- СПб: Питер Пресс, 1997.
- Шабалов Н.П. Неонатология.- СПб: Специальная литература, 1997.
- Chisholm CA, Heider AL, Kuller JA, et al. Prenatal diagnosis and perinatal management of fetal sacrococcygeal teratoma. Am J Perinatol. 1998 Aug;15(8):503-5.
- Dynio Honrubia. Sacrococcygeal teratoma. 1997.
- Inoue M, Kubota A. Antenatal diagnosis of sacrococcygeal teratoma with hydrops tefalis. 1994.
- Liu K.K. Lee KH.KU KW. Sacrococcygeal teratoma in children: a diagnostic challenge. 1994.
Переглянуто редакційною колегією I.B.I.S.: 12/12/2001
Синдром Коффіна-Сіріса
(Coffin-Siris Syndrome)
Наталія Олегівна Зимак-Закутня
Зав. медико-генетичною консультацією
Хмельницького клінічного пологового будинку
Синоніми: синдром п’ятого пальця. Синдром описаний у 1970 році Коффіном і Сірісом.
Основні діагностичні критерії:
- затримка психо-моторного розвитку;
- грубі риси обличчя;
- гірсутизм;
- гіпоплазія або відсутнсть нігтьової фаланги на 5 пальці руки чи ступні.
Додаткові діагностичні критерії:
- труднощі вигодовування у періоді новонародженості;
- вроджена вада серця;
- схильність до частих інфекцій;
- затримка прорізування зубів.
Клінічна симптоматика:
У більшості випадків синдрому Коффіна-Сіріса спостерігається помірна розумова відсталість (IQ від 40 до 70) або затримка розвитку, що представлені широким спектром від порушення мови, тонкої моторики до соціальної дезадаптації та затримки мовних функцій. Часті ознаки – м’язова гіпотонія, мікроцефалія, аномалія Денді-Уолкера, гіпоплазія мозочка, судоми. Зміни з боку очей проявляються міопією, астигматизмом, страбізмом, ністагмом. Спостерігається також різного ступеню вираженості зниження слуху.
Ектодермальні симптоми представлені гіпоплазією чи відсутністю нігтьової пластинки на 5-их пальцях китиць рук і ступнів, характерним малюнком росту волосся у вигляді поєднання рідкого волосся на голові та вираженого гіпертрихозу тулуба, аномальним ростом зубів, нефункціонуючими слізними протоками.
Лицьові прояви дещо схожі на такі при синдромі Гурлер, Корнелії де Ланге: грубі риси обличчя, широкий ніс, великий рот, широкі губи, макроглосія, пласке перенісся, широкі латерально розташовані брови, довгі вії, короткий фільтр.
Можливі спінальні аномалії у вигляді “сакральної ямки”, сколіозу, кифозу, spina bifida. Тулуб короткий, характерна затримка кісткового віку.
Асоційовані вади:
- вроджені вади серця, зокрема дефекти міжшлуночкової та міжпередсердної перетинок, тетрада Фалло, відкрита артеріальна протока;
- генітальні аномалії: крипторхізм, гіпоспадія, аплазія матки;
- аномалії нирок: гіпоплазія, злиття, подвоєння;
- пахові, пупкові кили;
- гіпермобільність суглобів;
- затримка внутрішньоутробного розвитку (середня маса при народженні при середньому терміні гестації 39 тижнів – 2600 г).
Лабораторні дані:
Лабораторна верифікація синдрому Коффіна–Сіріса не розроблена. Діагностика базується на підставі клінічних ознак та параклінічних обстежень з метою виявлення асоційованих вад розвитку.
Тип успадкування:
В оригінальному описі передбачався аутосомно-рецесивний тип успадкування. Сучасні уявлення виявляють аутосомно-домінантний тип успадкування.
Диференційна дагностика:
Синдроми Коффіна-Лоурі, Корнелії де Ланге, Гурлера, часткової трисомії короткого плеча 9 хромосоми, фетальний гідантоїновий синдром.
Генетичний ризик: для потомства пробанда – високий (50%).
Співвідношення статі: Ж4 : Ч1.
Лікування та нагляд:
Симптоматичне. Беручи до уваги спектр поєднаних аномалій, у неонатальному періоді рекомендується проведення ультразвукового обстеження нирок, серця з доплерокардіографією, обстеження головного мозку (нейросонографія, комп’ютерна томографія, магнітно-ядерний резонанс), офтальмологічне обстеження, дослідження шлунково-кишкового тракту, каріотипування для виключення подібних хромосомних синдромів.
Прогноз:
Щодо тривалості життя визначається поєднаними аномаліями, щодо затримки розумового розвитку несприятливий.
Пренатальна діагностика:
Специфічна – не розроблена. Пренатальна ультразвукова діагностика дозволяє виявити вади розвитку сечостатевої, серцево-судинної системи, затримку внутрішньоутробного розвитку плоду.
Номер з каталогу МІМ:
135900 Coffin-Siris Syndrome, CSS.
Література:
- Bonioli, E.; Palmieri, A.; Bertola, A.; Bellini, C.: Autosomal recessive mode of inheritance of a Coffin-Siris like syndrome. Genet. Counsel. 6: 309-312, 1995.
- Bonneau, D.; Berthier, M.; Oriot, D.; Munnich, A.: Coffin-Siris syndrome with normal plasma biotinidase activity. Europ. J. Pediat. 150: 687 only, 1991.
- Burlina, A. B.; Sherwood, W. G.; Zacchello, F.: Partial biotinidase deficiency associated with Coffin-Siris syndrome. Europ. J. Pediat. 149: 628-629, 1990.
- Carey, J. C.; Hall, B. D.: The Coffin-Siris syndrome: five cases including two siblings. Am. J. Dis. Child. 132: 667-671, 1978.
- Coffin, G. S.; Siris, E.: Mental retardation with absent fifth fingernail and terminal phalanx. Am. J. Dis. Child. 119: 433-439, 1970.
- DeBassio, W. A.; Kemper, T. L.; Knoefel, J. E.: Coffin-Siris syndrome: neuropathologic findings. Arch. Neurol. 42: 350-353, 1985.
- deJong, G.; Nelson, M. M.: Choanal atresia in two unrelated patients with the Coffin-Siris syndrome. Clin. Genet. 42: 320-322, 1992.
- Franceschini, P.; Silengo, M. C.; Bianco, R.; Biagioli, M.; Guala, A.; Lopez Bell, G.: The Coffin-Siris syndrome in two siblings. Pediat. Radiol. 16: 330-333, 1986.
- Gorlin, R. J.: Lapsus–caveat emptor: Coffin-Lowry syndrome vs Coffin-Siris syndrome–an example of confusion compounded. (Letter) Am. J. Med. Genet. 10: 103-104, 1981.
- Haspeslagh, M.; Fryns, J. P.; van den Berghe, H.: The Coffin-Siris syndrome: report of a family and further delineation. Clin. Genet. 26: 374-378, 1984.
- Levy, P.; Baraitser, M.: Coffin-Siris syndrome. J. Med. Genet. 28: 338-341, 1991.
- Mattei, J. F.; Laframboise, R.; Rouault, F.; Giraud, F.: Coffin-Lowry syndrome in sibs. Am. J. Med. Genet. 8: 315-320, 1981.
- Qazi, Q. H.; Heckman, L. S.; Markouizos, D.; Verma, R. S.: The Coffin-Siris syndrome. J. Med. Genet. 27: 333-336, 1990.
- Rabe, P.; Haverkamp, F.; Emons, D.; Rosskamp, R.; Zerres, K.; Passarge, E.: Syndrome of developmental retardation, facial and skeletal anomalies, and hyperphosphatasia in two sisters: nosology and genetics of the Coffin-Siris syndrome. Am. J. Med. Genet. 41: 350-354, 1991.
- Richieri-Costa, A.; Monteleone-Neto, R.; Gonzales, M. L.: Coffin-Siris syndrome in a Brazilian child with consanguineous parents. Rev. Brasil. Genet. IX: 169-177, 1986.
- Senior, B.: Impaired growth and onychodysplasia: short children with tiny toenails. Am. J. Dis. Child. 122: 7-9, 1971.
- Swillen, A.; Glorieux, N.; Peeters, M.; Fryns, J.-P.: The Coffin-Siris syndrome: data on mental development, language, behavior and social skills in children. Clin. Genet. 48: 177-182, 1995.
- Tunnessen, W. W.; McMillan, J. A.; Levin, M. B.: The Coffin-Siris syndrome. Am. J. Dis. Child. 132: 393-395, 1978.
- Weiswasser, W. H.; Hall, B. D.; Delavan, G. W.; Smith, D. N.: Coffin-Siris syndrome: two new cases. Am. J. Dis. Child. 125: 838-840, 1973.
Переглянуто редакційною колегією I.B.I.S.: 27/01/2003
|
|
|
|
|
|





