
Пентада Кантрелла
(Pentalogy of Cantrell)
Ірина Ткач
Акушер-гінеколог
Зарічненської ЦРЛ Рівненської області
Включення:
Торако-абдомінальна ектопія серця, торакоабдомінальний синдром, синдром Кантрелла, синдром Кантрелла-Геллера-Равіча, перитонео-перикардіальна діафрагмальна кила.
Визначення:
Пентада Кантрелла – це комплекс вроджених вад розвитку дизрафічного ряду. Повний синдром характеризується двома головними дефектами: ектопією серця і дефектом черевної стінки (частіше у вигляді омфалоцеле, хоча може також мати місце розходження м’язів черевної стінки). Вперше синдром описаний Кантреллом у 1958 році, спостерігається спорадично.
Основні діагностичні критерії:
Діагноз повного синдрому вимагає п’ятьох критеріїв, описаних Кантреллом, але спостерігаються і неповні форми. Дефекти грудини можуть виявлятися у вигляді відсутності мечоподібного паростка, повного розщеплення грудини, її вкорочення аж до повної відсутності грудини. Черевний дефект може бути у вигляді широкого розходження прямих м’язів живота або великого омфалоцеле. Найбільш поширені інтракардіальні вади проявляються у вигляді міжпередсердних та міжшлуночкових дефектів і тетради Фалло.
Відносно частоти аномалій, що зустрічаються при синдромі Катрелла, то абдомінально-стернальні дефекти зустрічаються в 74,5%; омфалоцеле в 59,4%; дефект діафрагми в 56,8%; дефект перикарда в 41,8%. Що стосується серцевих аномалій, то вони зустрічаються з частотою 83%, з них найчастіше відслідковуються інтравентрикулярні дефекти, дивертикул шлуночка, шлуночкові аневризми, стеноз легеневої артерії, декстрокардія, аномальне впадіння верхньої порожнистої вени, аномальне відходження легеневої артерії та відкрита артеріальна протока, інколи тетрада Фалло.
Рентгенівське дослідження може виявити дефекти грудини, декстрокардію та інші відхилення.
Магнітно-резонансна томографія дає можливість підтвердити наявність тих чи інших аномалій, а також дослідити серцево-судинну гемодинаміку.
Асоційовані аномалії:
Є повідомлення і про інші дефекти, а саме: розщілину піднебіння і губи, кистозну гігрому, клишоногість, вади формування грудини, транспозицію великих судин, відкриту артеріальну протоку, дефекти міжшлуночкової перегородки, дивертикул лівого шлуночка, гіпоплазію легень, мальротацію, гідроцефалію, аненцефалію, гіпоспадію та інші.
Диференційний діагноз:
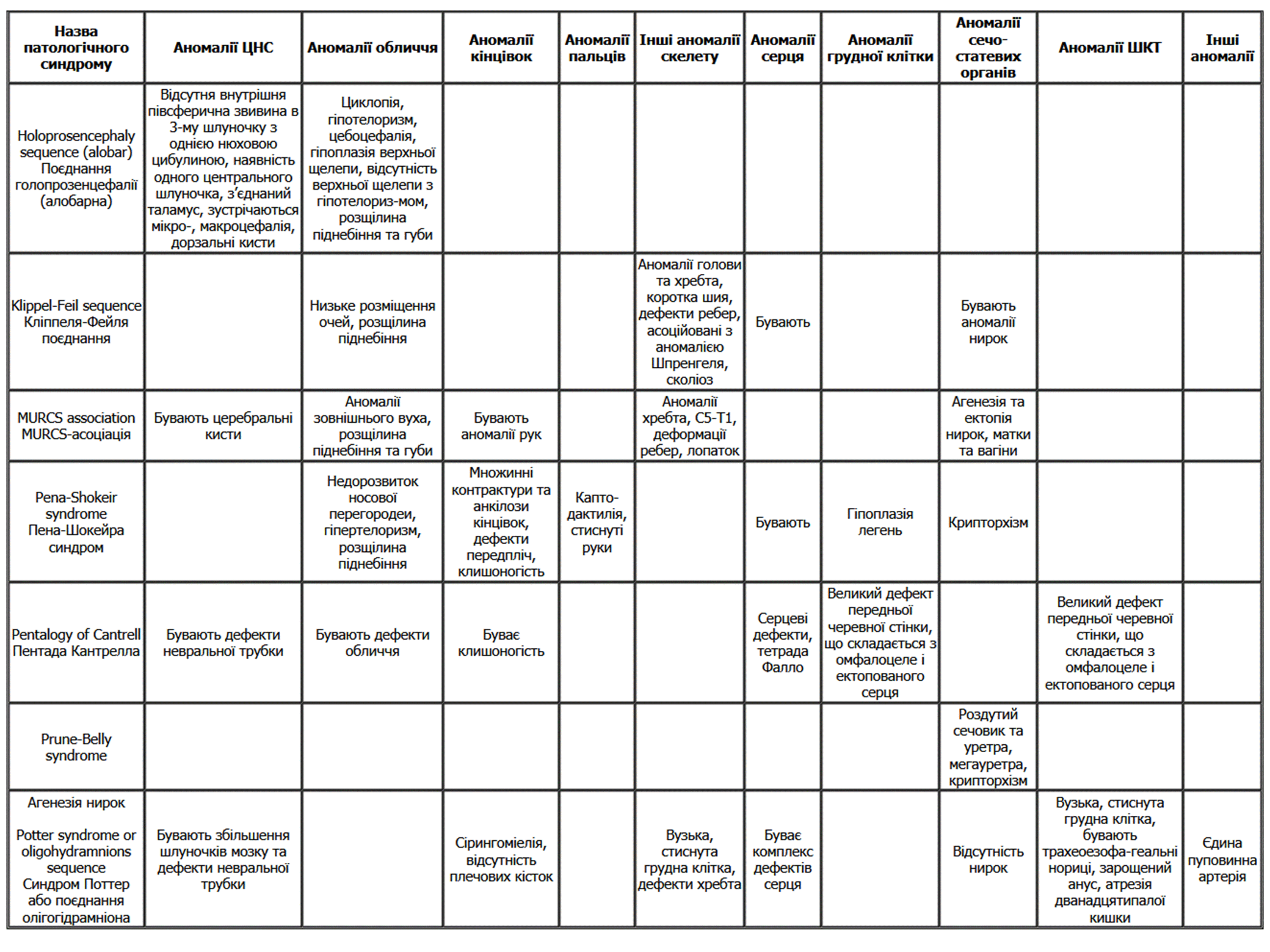
Диференційний діагноз слід проводити з ізольованою ектопією серця, ізольованим дефектом черевної стінки, ізольованим омфалоцеле, ізольованою тетрадою Фалло та іншими патологічними станами, викладеними у таблиці 1.
Етіологія:
Невідома. Іноді пов’язується з хромосомними аномаліями. Описані асоціації синдрому з трисомією 18, трисомією 13 і синдромом Тернера. Є дані, які підтверджують виникнення хвороби при успадкуванні Х-зчепленого гену (генний локус Xq25-q26.1).
Патогенез:
Основною причиною є порушення, пов’язане з розвитком сегменту мезодерми між 14 і 18 днями після запліднення, коли вісцеральна і корпоральна мезодерми розділяються. Грудна, черевна стінка, перикард і частина діафрагми є похідними мезодерми тіла, у той час як міокард є результатом поділу клітин органної мезодерми. Бічні мезодермальні клітини мігрують до серединної лінії зародкового диска і змикаються попереду. Якщо цього не відбувається, то залишаються дефекти черевної стінки, грудини, діафрагми і перикарду.
Порушення під час розподілу мезодерми призводять до порушень, які вкладаються в пентаду Катрелла.
Поширеність:
Дуже рідкісний. Зустрічається спорадично. Менше, ніж про 90 випадків було повідомлено в літературних джерелах. Зустрічається, за даними літератури, з частотою 5,5 випадків на 1 млн. живонароджених, причому переважно у чоловічої статі.
Пренатальна діагностика:
Можливість виявлення даних аномалій при сонографічному дослідженні залежить від вираженості, величини та сукупності вад.
Навіть при народженні встановити повну ступінь синдрому важко, оскільки дефект грудини може бути незначним і без істинної ектопії серця.
Прогноз:
За даними літератури відомо про виживання в 20% випадків. Це стосувалось випадків з помірними дефектами і неповним вираженням синдрому.
Повний прогноз здається несприятливим і цілком залежить від величини стернальних та вентральних дефектів.
Лікування:
Після народження дитини втручання з приводу ліквідації омфалоцеле не повинно відтягуватись. Відновлення дефектів грудини, діафрагми та перикарду може бути проведене одночасно.
Часто хірургічне втручання виконати важко або взагалі нереально внаслідок гіпоплазії грудної клітки і неможливості ліквідувати ектопію серця. Деякі новонароджені мають виражену дихальну недостатність, часто вторинну внаслідок легеневої гіпоплазії. Розпізнавання й ліквідація будь-яких інтракардіальних вад важливі, оскільки вони є основною причиною смерті в постнатальному періоді.
Номер з каталогу МІМ:
313850 Thoracoabdominal Syndrome; THAS.
Література:
- Пренатальная диагностика врожденных пороков развития плода /Р. Ромеро, Дж. Пилу, Ф. Дженти, А. Гидини, Дж.С. Хоббинс.- М.: Медицина.- 1994.- C. 222-223.
- Benacerraf BR. Ultrasound of fetal syndromes. Churchill Livingstone 1998:267-269.
- Thoracoabdominal Syndrome. OMIM #313850.
Переглянуто редакційною колегією I.B.I.S.: 27/08/2003
|
|
|
|
|
|





