
Serhiy
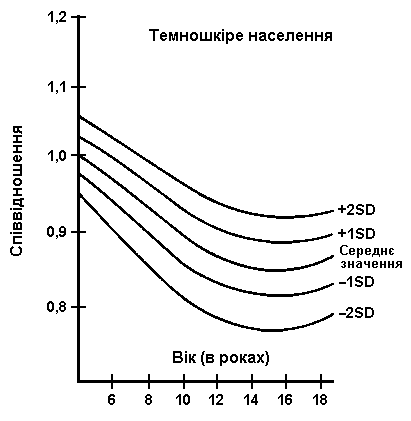
Співвідношення верхнього та нижнього сегментів тіла
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”.

Співвідношення верхнього сегменту до нижнього сегменту для представників білої раси подано зверху, для темношкірих – внизу. На діаграмах поданий середній показник та 2 стандартні відхилення. Дані були отримані в 1959 році після обстеження 2104 школярів з м. Балтимор (1015 білих, 1089 чорних). McKusick VA: Heritable Disorders of Connective Tissue, CV Mosby, 1972.
Синдром Марфана – співвідношення верхнього та нижнього сегментів тіла
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”.

Шкали співвідношення верхнього та нижнього сегментів тіла осіб з синдрмом Марфана накладаються на криві нормального розвитку. Крапки показують середні показники для осіб європеоїдної раси, згрупованих по віку з інтервалом в 1 рік +/- одне стандартне відхилення. Суцільна лінія показує середній розвиток, а пунктирна – два стандартних відхилення нижче середнього показника (дивіться сторінку “Співвідношення верхнього та нижнього сегментів тіла” в розділі постнатального розвитку). Дані збирались під час обстеження пацієнтів з синдромом Марфана в університеті Джона Хопкінса. Pyeritz RE: Marfan syndrome, in Principles and Practices of Medical Genetics, Churchill Livingstone, 1983. Pyeritz RE et al: Growth and Anthropometrics in the Marfan syndrome, in Endocrine Genetics and Genetics of Growth, Alan R. Liss, 1985.
Синдром Марфана – зріст та вага дівчат
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”.
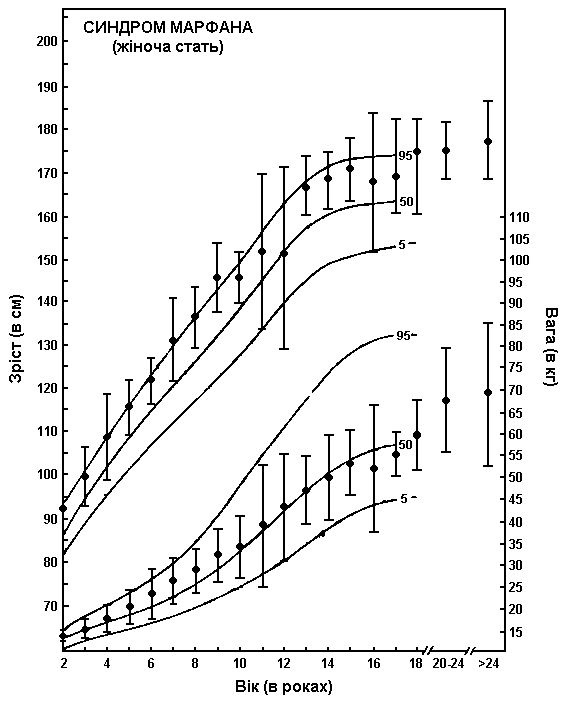
Шкали зросту та ваги дівчат з синдромом Марфана накладаються на криві нормального розвитку (5-й, 50-й та 95-й перцентилі). Для створення діаграми використовувались дані довготривалого перехресного обстеження 200 дівчат європеоїдної раси, хворих на синдром Марфана. Пацієнтів не лікували гормонами. Риски показують ± одне стандартне відхилення. Pyeritz RE: Marfan syndrome, in Principles and Practice of Medical Genetics, Churchill Livingstone, 1983.
Синдром Марфана – зріст та вага хлопчиків
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”.
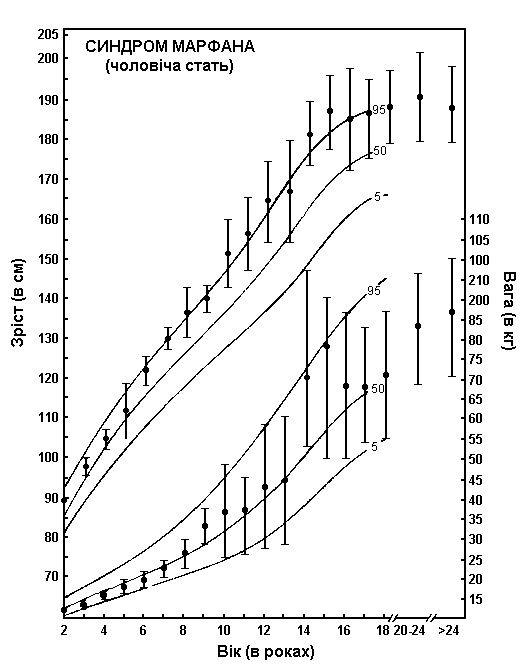
Шкали зросту та ваги хлопчиків з синдромом Марфана накладаються на криві нормального розвитку (5-й, 50-й та 95-й перцентилі). Для створення діаграми використовувались дані довготривалого перехресного обстеження 200 чоловіків європеоїдної раси, хворих на синдром Марфана. Пацієнтів не лікували гормонами. Риски показують ± одне стандартне відхилення. Pyeritz RE: Marfan syndrome, in Principles and Practice of Medical Genetics, Churchill Livingstone, 1983.
Синдром Марфана
(Marfan Syndrome)
Сергій Федорович Лапченко
Спеціаліст з інформаційного забезпечення
Українсько-Американської Програми запобігання вродженим вадам розвитку
Синдром Марфана є одним із широко розповсюджених спадкових захворювань сполучної тканини. Він зустрічається однаково часто у чоловіків і жінок усіх расових та етнічних груп.
Синдром названий іменем доктора Антуана Марфана, який в 1896 році вперше описав 5-річного пацієнта з довгими тендітними кінцівками і пальцями та іншими скелетними аномаліями. Симптоми синдрому Марфана можуть бути м’якими або дуже вираженими, вони можуть проявлятися як одразу після народження, так і значно пізніше. Хвороба іноді спричиняє раптову смерть у дорослих, які навіть і не підозрювали, що є хворі.
Що таке синдром Марфана?
Це широкий спектр аномалій серця, кровоносних судин, легень, очей, кісток та зв’язок. Він об’єднує більше 100 спадкових захворювань сполучної тканини. Хворі завжди мають високий ріст, є тендітними та мають дуже гнучкі суглоби. Руки і ноги можуть бути незвичайно довгими у порівнянні з торсом. Хребет може бути викривлений (сколіоз), часто хворі мають випираючу або запалу грудну кістку. Обличчя може бути видовженим і вузьким, піднебіння – високим, а зуби можуть рости дуже густо.
Майже завжди страждають серце і судини. Два або й три серцевих клапани, які відповідають за те, щоб кров текла через серце в певному напрямку, є збільшені і розхитані. Під час серцевих скорочень вони пропускають певну кількість крові в зворотньому напрямку, викликаючи серцевий шум (аномальний звук, який можна почути через стетоскоп).
Майже завжди є певний ступінь ураження найбільшої людської артерії – аорти. Серце з великою силою виштовхує кров в аорту, яка розгалужується наче дерево, щоб рознести збагачену киснем кров по всьому тілі. Слабка аорта поступово розтягується і рветься в найтонших місцях, через які кров просочується в грудну і черевну порожнини. Раптові, великі розриви призводять до внутрішньої кровотечі і смерті, як це сталося в 1986 році із знаменитою волейбольною зіркою, членом олімпійської команди США Фло Хаймен. На додачу, хворі з синдромом Марфана схильні до раптового колапсу легень.
У 50% людей з синдромом Марфана кришталик ока знаходиться поза центром. Втім, незалежно від місцезнаходження кришталика, майже завжди діагностується короткозорість. У хворих часом може початися відшарування сітківки, що призводить до сліпоти.
Яка причина виникнення синдрому Марфана?
В 1991 році вчені довели, що хвороба виникає через дефектний ген в 15 хромосомі. В нормі цей ген відповідає за виробництво спеціального протеїну (фібріліну), який робить сполучну тканину міцною і еластичною. Особливо багато фібріліну знаходиться в аорті, зв’язках, які тримають кришталик ока на місці, і в кістках. Очевидно, люди з синдромом Марфана мають в цих тканинах мало фібріліну (або ж він змінений), що змушує тканини розтягуватися через нездатність витримувати нормальний тиск.
Дефектний ген звичайно передається від одного з батьків, який має цю хворобу. Це – “домінантний” ген, що означає, що кожна дитина такого батька (чи матері) має 50-60% шансів захворіти. В 25% випадків відбувається нова мутація в сперматозоїді або яйцеклітині, в результаті чого народжується хвора дитина у повністю здорових батьків.
Синдромом Марфана, як і любою іншою спадковою хворобою, неможливо заразитися від хворої людини.
Чи можливо діагностувати синдром Марфана на ранній стадії розвитку?
На жаль, сьогодні немає точного тесту, який би дозволив діагностувати синдром до того, як з’являться потенційно небезпечні для життя симптоми, що дало б можливість вчасно розпочати лікування і врятувати багатьох людей від смерті.
Проте, в деяких сім’ях, де хворіють синдромом Марфана вже кілька поколінь, генетичне тестування можливе й сьогодні.
Деякі лікарі, добре знайомі з хворобою, можуть діагностувати її після детального огляду серця, аорти, очей та інших частин тіла. На жаль, через велику варіабельність симптомів і різну ступінь проявів хвороби, зробити це буває дуже нелегко.
Відкриття гена, що спричиняє синдром, повинно допомогти вченим в майбутньому створити точний тест для діагностики синдрому Марфана.
Як лікують синдром Марфана?
Найбільшу небезпеку для хворого створює раптовий розрив аорти. Це може статися під час нормального проходження крові через слабку аорту або через підвищену фізичну чи емоційну активність, що викликають підвищений тиск крові в судинах. Тому дітям і дорослим з синдромом Марфана рекомендують уникати тяжких фізичних вправ, занять спортом, підйому тяжких речей. Однак, таким людям можна їздити на велосипеді або плавати, певна річ, якщо це дозволяє лікар.
Регулярні медичні огляди, тестування серця і аорти допомагають боротися з симптомами хвороби до того, як вони становлять смертельну небезпеку. Часом призначаються препарати, що знижують тиск крові і зменшують частоту серцебиття (бета-блокатори). Дослідження доводять, що вони уповільнюють процес розширення аорти і запобігають її розривам.
Можливе також оперативне лікування, коли ушкоджений аортальний клапан з частиною аорти заміняється штучним клапаном, приєднаним до трубки довжиною 10 см. Після такої операції пацієнту призначають антитромботичну терапію, бо кров має тенденцію до утворення тромбів (згортається) в місці контакту з штучними клапанами.
Через дефекти серцевих клапанів, люди з синдромом Марфана схильні до інфекцій, які розвиваються в цих клапанах. Тому вони повинні приймати антибіотики перед любим хірургічним втручанням, включаючи зубне лікування, щоб не дати можливості бактеріям попасти в кров.
Лікувальна фізкультура, застосування спеціальних корсетів або хірургічне втручання можуть допомогти виправити викривлення хребта. Стоматолог-ортодонтист допоможе справитися із зубними проблемами, а офтальмолог – з проблемами очей. Відшарування сітківки часом може бути виправлене за допомогою безкровної лазерної хірургії.
Хворі на синдром Марфана вагітні жінки вважаються групою підвищеного ризику незалежно від того, чи мають вони симптоми розширення аорти. Справа в тому, що, по-перше, є 50% ризик передати синдром дитині у спадок, по-друге, можливість розриву аорти під час вагітності і родів значно збільшується.
За винятком рідких випадків, коли дитина народжується з дуже тяжкою формою синдрому Марфана, рання діагностика і відповідне лікування допомагають хворим жити довше і зручніше.
Чи можливо запобігти виникненню синдрома Марфана?
Сьогодні неможливо попередити чи вилікувати синдром Марфана. Рання діагностика може лише запобігти серйозним ускладненням хвороби. Генетичне консультування дозволяє свідомо приймати рішення щодо майбутньої або існуючої вагітності.
Всі хворі синдромом Марфана люди і їх сім’ї повинні знати, які тяжкі можуть бути ускладнення і яки методи їх лікування існують в сучасній медицині.
Переглянуто редакційною колегією I.B.I.S.: 05/02/2002
Дивіться також:
- Синдром Марфана (інформація для спеціалістів)
- Синдром Марфана (коротка інформація)
- Синдром Марфана (діаграми розвитку дітей)
Синдром Марфана
(Marfan Syndrome)
Галина Володимирівна Скибан
Зав. поліклінікою науково-дослідного інституту
педіатрії, акушерства і гінекології (НДІ ПАГ), м. Київ
Основні діагностичні критерії:
Високий зріст, арахнодактилія, доліхостеномелія, гіперрухливість суглобів, сколіоз, підвивих кришталика, аневризма аорти.
Клінічна характеристика:
Для хворих синдромом Марфана характерними є високий зріст, диспропорційне довгі кінцівки і пальці, деформація передньої стінки грудної клітки, помірна рухомість суглобів, деформації хребта (сколіоз або грудний лордоз) і високе готичне піднебіння з патологією зубів і частими скелетними аномаліями. Можуть спостерігатись також міопія, двосторонній підвивих кришталика, іридодонез, сферофакія, мікрофакія, відшарування сітківки, гетерохромія райдужки, мегалокорнеа і голубі склери. Серед патологічних змін серцево-судинної системи мають місце ураження великих судин і серця: пролапс мітрального клапана, мітральна регургітація, дилятація аорти, аортальна регургітація, аневризма аорти. Нерідко спостерігаються стегнові, пахові і діафрагмальні кили, гіпоплазія м’язів, м’язова гіпотонія, нефроптоз, емфізема легенів, пневмоторакс.
Біохімічне визначається аномалія первинної структури колагену синтезу гіалуронової кислоти, що характерно для мутації альфа-2 гену колагена І, а також інших колагенових генів.
У більшості випадків причиною захворювання є мутація гена фібриліну-І, локалізованого на хромосомі 15 (q15q15 – q21.3).
Ускладнення:
Нестабільність суглобів, тяжкі деформації грудної клітки і захворювання легень, втрата зору, аортальна і мітральна регургітація, раптова смерть, бактеріальний ендокардит.
Етіологія:
Аутосомно-домінантне успадкування з повною пенетрантністю і високо варіабельною експресивністю. Гомозиготний стан є летальним.
Патогенез:
Важливу роль відіграє порушення синтезу глікопротеїну, фібриліну, основного компоненту мікрофібрію.
Популяційна частота:
1 : 10000. Біля 15-30% – нові мутації.
Профілактика:
Медико-генетичне консультування.
Диференційний діагноз:
Гомоцистинурія, арахнодактилія контрактурна вроджена, Елерса-Данлоса синдром.
Номер з каталогу МІМ:
154700 Marfan syndrome; MFS.
Література:
- С.И. Козлова, Н.С. Демикова, Е. Семанова, О.Е. Блинникова. Наследственные синдромы и медико-генетическое консультирование.- М.: Практика, 1996.- 416 с.
- Abraham P.A., Perioda A.J., Carnes W.H., Uitto J., Marfan synrome: demonstration of abnormal elastin in aorta. J.Clin.Invest. 1982;70:1245-1252.
- Buyse M.L. Birth Defects Encyclopedia: Center for Birth Defects Information Services, Dover, 1990:610-611.
- Gorlin R.J., Cohen M.M., Levin L.S. Syndromes of the Head and Neck, NY, Oxford: Oxford University Press, 1990:429-441.
- Jones K.L. Smith’s recognizable Patterns of Human Malformations, Philadelphia, London, Torornto: W.B.Saunders Company, 1988:472-475.
- Warkany J. Congenital malformations. Notes and Comments. Chicago: Year book medical publishers, 1971:850-857.
Переглянуто редакційною колегією I.B.I.S.: 05/02/2002
Дивіться також:
- Синдром Марфана (інформація для спеціалістів)
- Синдром Марфана (інформація для батьків)
- Синдром Марфана (діаграми розвитку дітей)
Синдром Марфана
(Marfan Syndrome)
Любов Степанівна Євтушок
Зав. медико-генетичною консультацією
Рівненського клінічного лікувально-діагностичного центру
Загальні відомості:
Синдром Марфана – це спадкове захворювання сполучної тканини, що успадкується як аутосомно-домінантна ознака. 25-30% випадків є наслідком нових мутацій. Основні прояви класичної форми включають скелетні, очні, кардіоваскулярні симптоми та зміни з боку центральної нервової системи (твердої оболонки). Синдром Марфана характеризується високою пенетрантністю та варіабельністю клінічних ознак. Діагностика синдрому грунтується на даних клінічного обстеження. Чітко визначені діагностичні критерії допоможуть відрізнити синдром Марфана від інших подібних порушень. Причиною захворювання є мутації в гені фібріліну (FBN1). Ген фібрілину локалізований на довгому плечі хромосоми 15 (15q21).
Клінічні прояви:
Основні клінічні прояви включають доліхостеномелію (непропорційно довгі руки та ноги), арахнодактилію, ектопію кришталика, пролапс мітрального клапану та дилатацію аорти. Зазвичай, пацієнти мають високий зріст, переважання розмірів нижнього сегменту тіла над верхнім, гіпермобільність суглобів рук, видовжене обличчя, високе піднебіння, порушення росту зубів, прогнатизм, деформації грудної клітки та сколіоз.
Два найбільш поширених кардіоваскулярних порушення: пролапс мітрального клапану та розширення висхідної аорти.
Очні прояви включають в себе підвивих кришталика або неправильне його розташування у 50-80% пацієнтів, вторинну міопію, відшарування сітківки, глаукому та запалення райдужної оболонки ока.
Може зустрічатися розширення твердої оболонки спинного мозку в попереково-крижовому відділі. В більшості випадків воно є асимптоматичним, хоч може спричиняти біль в нижній частині спини та загальну слабкість. Під час проведення комп’ютерної томографії або магнітно-резонансного сканування помітно розширення неврального каналу.
Інші клінічні прояви включають: полоси розтягнення на шкірі плечей, бокових поверхонь тулуба та поперекового відділу хребта; кили; спонтанний пневмоторакс; психоневрологічні порушення, такі як нездатність до навчання та концентрування уваги.
Підхід до діагностики:
Діагностика захворювання включає вивчення родинного анамнезу (родоводу), ретельний об’єктивний огляд (включаючи вимірювання пропорцій тіла), детальне офтальмологічне обстеження, включаючи огляд через щілинну лампу, ехокардіографію, скрінінг на сколіоз.
Диференційний діагноз:
Необхідно виключити: гомоцистінурію, контрактурну арахнодактилію, декілька варіантів синдрому Елерса-Данлоса, родинні випадки пролапсу мітрального клапана.
Поради щодо лікування:
Специфічної терапії для даної вади немає. Усі зусилля спрямовуються на попередження симптомів, які можуть спричинити загрозу для життя або здоров’я. Ефективна програма передбачає динамічний нагляд багатьох спеціалістів, включаючи педіатра і терапевта, консультації кардіолога, окуліста, ортопеда, генетика.
Немовля та дитина:
Принаймні один раз на рік пацієнт повинен пройти повне медичне обстеження: дослідження серця і судин (ехокардіографія); скрінінг на сколіоз; в разі потреби – ортопедичний огляд (неправильна постава, деформація грудної клітки та сколіоз); обстеження парадонтолога (якщо є відставання у прорізуванні зубів або аномалії прикусу); профілактика бактеріальних ендокардитів; заборона певних видів діяльності, спрямована на захист аорти (наприклад, контактних видів спорту та ізометричних вправ); залучення до таких видів спорту, як плавання та велосипед; застосування бета-блокаторів (пропранолол або атенолол); в разі потреби – консультації психолога та психоневролога; генетична консультація для пацієнта та родини.
Юність та доросле життя:
Більшість рекомендацій, призначених для дітей, також придатні й для дорослих хворих із синдромом Марфана. Переважна більшість скелетних та очних проявів при синдромі Марфана не прогресують з віком. Найбільш складні та потенційно фатальні ускладнення асоціюються із кардіоваскулярними проявами. Смертність серед дорослих пацієнтів спричиняється ураженням кореня аорти з її прогресуючою дилатацією, наслідком якого може стати аневризма або розрив аорти. Тому наголос ставиться на спостереженні за кардіоваскулярною системою. Кратність лікарських оглядів залежить від складності клінічних проявів. Для синдрому Марфана середнього ступеня важкості (пролапс мітрального клапана із незначною дилатацією аорти) рекомендується щорічний огляд кардіолога і обов’язково ехокардіографія. Для складніших випадків дилатації аорти і регургітації крові рекомендується більш часте відвідування лікаря, ехокардіографія, комп’ютерна томографія та магнітно – резонансне сканування.
Вірогідність регургітації крові в аорті є незначною для пацієнтів, в яких діаметр кореня аорти становить від 3,6 см та менше, проте стає значною, якщо діаметр її дорівнює 6 см та більше. Хірургічна корекція розширення чи аневризми аорти і одночасно заміна аортального клапана має велике значення для виживання пацієнтів із синдром Марфана. Результати 10-річних досліджень свідчать, що рівень виживання хворих становить понад 75%.
Сколіоз має схильність до прогресування протягом підліткового прискорення росту. Викривлення розміром більше ніж 10 градусів необхідно лікувати спеціальними вправами та фізіотерапією. Хірургічне втручання стає необхідним, коли викривлення хребта прогресує і перевищує 40 градусів. Зріст дорослих жінок, які проходили курси терапії естрогенами перед менархе, що сприяло пубертації та закриттю епіфізів, був нижчим. Проте немає переконливих доказів того, що прояви сколіозу зменшуються при естрогенній терапії у підлітковому віці.
Деформації грудної клітки спостерігаються у 2/3 пацієнтів із синдромом Марфана, однак хірургічну корекцію слід проводити лише у випадках кардіопульмонарних ускладнень, а не заради косметичного ефекту. Деформація грудної клітки може рецидивувати, якщо операція була проведена в дитячому віці. Тому хірургічну корекцію треба відкласти до дозрівання скелету.
1 раз на рік необхідна консультація офтальмолога, що має досвід у лікуванні захворювань сполучної тканини. Лікування скероване на корекцію амбліопії, підвивиху кришталика, аномалій передньої камери, катаракти, глаукоми та відшарування сітківки. Покращання зору більше, як 20/40 можна досягти без хірургічного втручання у більш як 90% пацієнтів. Підвивих кришталика рідко вимагає його видалення, виключення становлять випадки, коли корекція зору неможлива або коли кришталик є зміщеним до передньої камери. Вдосконалені методи інтраокулярної хірургії зменшили ризик постопераційного відшарування сітківки до 5-10%.
Щодо підвищеного ризику виникнення пневмотораксу, пацієнтам із синдромом Марфана слід уникати різких змін атмосферного тиску (це може відбуватися при глибоководному зануренні та при польотах на негерметизованих літаках).
Вагітність:
Жінки із синдромом Марфана повинні знати як про 50% ризик виникнення хвороби для їх дітей, так і про можливі кардіоваскулярні ускладнення для них самих протягом вагітності. Фактори ризику під час вагітності: розширення аортального кореня понад 40 мм; аортальна регургітація; розрив аорти; пролапс мітрального клапану; дисфункція лівого шлуночка, особливо дилатаційна кардіоміопатія; будь-які інші структурні кардіоваскулярні аномалії; родинна схильність до аортальних розривів; ризик є найбільшим у другій половині вагітності та протягом кількох місяців після пологів.
Під час вагітності призначається обстеження серця та судин (в тому числі ехокардіографія) через кожні 6-8 тижнів. В цей час можна призначати бета-блокатори, які не підвищують тератогенного ризику для плода. Однак, при будь-якому підході пологи будуть гемодинамічним стресом для матері.
Життєвий прогноз:
Протягом останніх 20 років тривалість життя пацієнтів із синдромом Марфана збільшилась на 25%. Це відбувається завдяки досягненням кардіоваскулярної хірургії та своєчасному профілактичному хірургічному втручанню.
Медико-генетичне консультування:
Хворий має 50% ризик передати ген будь-кому з своїх нащадків. Якщо синдром Марфана виявляється у дитини, в родоводі якої не було вказівок на синдром Марфана, її батьків необхідно ретельно обстежити на наявність ознак синдрому. Якщо фенотип обох батьків в нормі, можна стверджувати, що ризик хвороби для їхніх майбутніх дітей є низьким. Дуже мало родоводів, документально свідчать про мозаіцизм в гермінативних тканинах.
Батьків слід інформувати про мінливість внутришньосімейних проявів: діти можуть бути уражені більше або менше, ніж їхні батьки.
Синдром Марфана в дитячому віці:
Хоча перші випадки синдрому Марфана були зареєстровані у дітей та підлітків, цю ваду важко діагностувати в ранньому віці. З іншого боку, у деяких дітей виявляються важкі клінічні прояви з раннього дитячого або неонатального віку. Фенотипічні прояви включають характерний старший, як для даного віку, вираз обличчя з глибоко посадженими очима, високе піднебіння, арахнодактилію, деформації грудної клітки, згинальні контрактури, пласкі ступні, вади серцевих клапанів, дилатацію кореня аорти та ектопію кришталика.
Діагностичні критерії синдрому Марфана
Другорядні критерії:
Для того, щоб родовід враховувався при визначенні діагнозу, має бути наявним принаймні один з основних критеріїв. |
Другорядні критерії:
Для оцінки кардіоваскулярної системи має бути присутнім основний критерій або один з другорядних. |
Основні критерії – необхідна наявність принаймні 4 з наступних проявів:
Другорядні критерії:
Мають бути наявними 2 основних критерії або один основний та 2 другорядних |
Другорядні критерії:
Для легенової системи достатньо наявності одного критерію. |
Другорядні критерії:
Для шкіри та її похідних важлива наявність будь-якого з критеріїв. |
Другорядні критерії:
Для діагностування аномалій ока вимагається наявність принаймні двох другорядних критеріїв. |
Для родинних випадків:
|
Номер з каталогу МІМ:
154700 Marfan syndrome; MFS.
Література:
- Borges M. Lecture delivered for medical students (May, 1999, Mobile, Alabama).
- Devreux RB, Roman MJ. Aortic disease in Marfan’s syndrome. N Engl J Med 1999;Apr 29;340(17):1358-9.
- Felbinger DM, Akers MC. Marfan’s syndrome in pregnancy: implications for advanced practice nurses. AACN Clin Issues 1998;Nov;9(4):563-8.
- Gielerak G, Wierzbicki P. Marfan’s syndrome: diagnosis and treatment. Pol Merkuriusz Lek 1998;Aug;5(26):98-100.
- Marfan syndrome; MFS. OMIM. #154700.
- Pyeritz RE. The Marfan syndrome. Annu Rev Med 2000;51:481-510.
- Rakov AL, Kulyga VN, Denisov SG, Karpalov VT. The early diagnosis of the Marfan syndrome in servicemen. Voen Med Zh. 1999;Jan;320(1):37-40.
- Tambeur L, David TE, UngerM, Armstrong S, Ivanov J, Webb G. Results of surgery for aortic rooy aneurismin patients with the Marfan syndrome. Eur J Cardiothorac Surg 2000;Apr;17(4):415-9.
Переглянуто редакційною колегією I.B.I.S.: 15/02/2002
Дивіться також:
- Синдром Марфана (коротка інформація)
- Синдром Марфана (інформація для батьків)
- Синдром Марфана (діаграми розвитку дітей)
Спадкові хвороби сполучної тканини при вагітності
(Congenital anomalies of the conjunctive tissue during the pregnancy)
І.І. Миронюк
Лікар акушер-гінеколог
Пологовий будинок №2, м. Рівне
Синдром Елерса-Данлоса
Синдром Елерса-Данлоса включає в себе більше 8 різних нозологічних форм:
- І тип – важкий, “класичний”, характеризується підвищеною розтяжністю шкіри, збільшенням об’єму рухів в суглобах, ламкістю тканин, геморрагічним діатезом. Пролапс мітрального клапана з або без пролапсу трьохстулкового клапана. Ймовірність народження хворою жінкою хворої дитини становить 50%.
- IІ тип – проміжний, успадковується аутосомно-домінантно.
- IІI тип – доброякісна гіпермобільність суглобів. У віці 30-40 років розвивається остеоартрит. Успадковується аутосомно-домінантно.
- IV тип – васкулярний тип, характеризується розривами судин, спонтанною перфорацією кишечника, утворенням гематом, аутосомно-домінантним типом.
- V тип – характеризується рецесивним зчепленим з Х-хромосомою типом успадкування. Клінічні ознаки подібні до ІІ типу.
- VI тип – крихкість тканин ока, важкий сколіоз, аутосомно-рецесивний тип успадкування.
- VII тип – ортопедичні порушення внаслідок повторних вивихів суглобів; аутосомно-рецесивний тип успадкування.
- VIII тип – важкий, аутосомно-домінантний тип успадкування.
- IX тип – Х-зчеплений рецесивний, характерні екзостози потиличної кістки.
- X тип – аутосомно-рецесивний тип успадкування; гіпермобільність суглобів, петехії.
- XI тип – аутосомно-домінантний тип успадкування; схильність до крововиливів.
Вагітність при синдромі Елерса-Данлоса І типу є ризикованою як для матері, так і для плода. У хворих жінок під час вагітності можуть бути:
- підвищена схильність до крововиливів в м’які тканини;
- кили черевної стінки, пахові, пупкові, стегнові;
- варикозне розширення вен обох нижніх кінцівок і статевих органів.
Описані випадки розривів великих судин під час вагітності, а також важкі післяпологові кровотечі, кровотечі з місця епізіотомії і гематоми промежини. Післяпологова кровотеча може бути настільки стійка, що потребуватиме гістеректомії.
Інші ускладнення синдрому Елерса-Данлоса І типу:
- розходження лонного симфізу під час пологів з наступним тривалим дискомфортом;
- випадіння матки і повільне загоєння рани;
- розрив піхви і відрив сечового міхура;
- повільне загоєння місця епізіотомії і розрізу після кесарського розтину і навіть їх спонтанне збільшення. Всі накладені шви слід залишати вдвічі довшими звичайних. Це попереджує розходження країв рани;
- гематоми промежини;
- підвищений ризик розриву рубця на стінці матки під час пологів у жінок, яким проводився кесарський розтин. Повторний кесарський розтин слід починати до початку пологової діяльності.
Передчасні пологи внаслідок передчасного розриву навколоплідних оболонок (якщо дитина несе мутантний ген) чи внаслідок перерозтягнення шийки матки (неспроможність шийки; швидке згладжування шийки може спричинити стрімкі пологи).
Передчасний розрив навколоплідних оболонок настає між 30-32-35 тижнями вагітності. Є вказівки на меншу вагу у вражених дітей, підвищену гнучкість, схильність до розвитку вродженого вивиху кульшових суглобів.
В наш час діагностика І, ІІ, ІІІ типів синдрому Елерса-Данлоса пренатально неможлива, оскільки невідомі біохімічні порушення, що лежать в його основі. Ті варіанти синдрому, для яких відомий специфічний дефіцит ферменту чи порушення структури колагену, можна діагностувати пренатально.
Гінекологічні порушення у жінок з синдромом Елерса-Данлоса:
- випадіння матки;
- випадіння сечового міхура;
- кили черевної стінки;
- геморрагічний діатез призводить до метроррагії і меноррагії;
- більша ймовірність перфорувати матку у жінок, що використовують внутрішньоматкові спіралі.
Рекомендації хворим з синдромом Елерса-Данлоса:
- Жінкам, у яких раніше були передчасні пологи, можна рекомендувати ліжковий режим.
- Кесарський розтин слід проводити за строгими показами.
- Хворі на синдром Елерса-Данлоса потребують медико-генетичної консультації.
- У випадку хвороби батька доцільно ставити питання про штучне запліднення.
Цікаво, що у жінок з синдромом Елерса-Данлоса не утворюються смуги розтягнення на шкірі живота; не властиві їм і порушення серцево-судинної системи, у всякому випадку при відсутності сколіозу.
Дивіться також:
- Синдром Елерса-Данлоса (інформація № 1)
- Синдром Елерса-Данлоса (інформація № 2)
Синдром Марфана
Характеризується:
- Патологією скелету (перерозтягнення, сколіоз, деформація грудної клітки, довгі тонкі кінцівки).
- Патологією очей (підвивих кришталика, міопія, відшаруння сітківки).
- Патологією серцево-судинної системи (розширення і розшарування висхідної частини аорти, розширення аортального клапана, в’ялість мітрального клапану з мітральною регургітацією).
Популяційна частота: 0,04:1000.
Синдром успадковується як аутосомно-домінатна ознака. Якщо батьків хворого обстежити, то можна встановити, що синдром має сімейний характер. Але в деяких випадках, особливо, де старший за віком батько, синдром Марфана є результатом нової домінантної мутації.
Загрозливим для життя порушенням серцево-судинної системи при синдромі Марфана є кистозний медіальний некроз стінки аорти, який створює передумови до формування аневризми. Аортальне кільце розширюється, що призводить до регургігації аорти і недостатності лівого шлуночка.
За даними літератури, аневризма аорти у жінок молодших 40 років з синдромом Марфана в 50% випадків формується під час вагітності, бо тоді підвищена частота розшарування і розриву аневризми аорти. Періодом найбільшого стресу для аорти є ІІІ триместр, причому під час пологів і в післяпологовому періоді частота розриву аневризми аорти не підвищена. У хворих з синдромом Марфана при вагітності підвищується і частота розриву селезінкової артерії. Є повідомлення і про розшарування вінцевої артерії. Під час вагітності з успіхом проводили протезування аорти.
Серед вагітних жінок з синдромом Марфана найбільш загрозливі ті, у котрих до вагітності вже були зміни аортального клапана чи розширення аорти, тому що під час вагітності такого ж типу розширення може прогресувати до аневризми. Жінки без змін клапана менше схильні до розвитку аневризми. Ступінь ризику точно невідомий. На думку деяких авторів, смертність вагітних жінок з синдромом Марфана досягає 50%, але ця цифра здається дещо завищеною. Серед жінок, які завагітніли вже при ураженні клапана аорти, під час вагітності приблизно 10-20% гинуть внаслідок аортальної катастрофи. Ризик смерті під час вагітності для жінок без ураження аорти складає, ймовірно, менше 5%. Таким чином, при сформованому ураженні аорти краще уникати вагітності. Враховуючи, що ризик розвитку аневризми аорти з віком підвищується, жінкам з синдромом Марфана рекомендується планувати вагітність до 30 років.
Якщо у матері виражений сколіоз, крім загрозливого для життя розшарування аорти під час вагітності, можуть бути дихальні розлади. Тому, при наявності сколіозу і до вагітності, і під час вагітності необхідно слідкувати за функцією легень.
Жінки з синдромом Марфана не відрізняються особливою схильністю до ускладнень при кесарському розтині; рани у них заживають нормально. Якщо є уражений лівий передсердно-шлуночковий (мітральний) клапан, необхідно проводити профілактику ендокардиту антибіотиками. Вагітність при синдромі Марфана асоціюється з підвищеною частотою утворення кил і полос розтягнення. Артеріальна гіпертензія під час вагітності для жінок з синдромом Марфана не характерна. Але необхідно постійно слідкувати за артеріальним тиском і регулювати його, тому що гіпертензія підвищує ризик розриву аневризми аорти.
Ускладнень, специфічних для ураженого і для неураженого новонародженого чекати не приходиться. Уражені діти складають 50% від загальної кількості дітей (серед уражених дівчаток і хлопчиків порівну). Діагноз можна поставити вже в період новонародженості.
Порушення імплантації і безпліддя, ймовірно, не властиві синдрому Марфана. Діти, які народжені від жінок з синдромом Марфана уражені не важче, ніж діти, народжені від хворого батька. Є дані, що хворі з синдромом Марфана, зумовленого новою мутацією, вражені більш виражено, ніж у сімейних випадках.
Гінекологічні порушення: слід відмітити, що дівчатка з синдромом Марфана в пубертатний період вступають нормально. За даними літератури, рекомендували призначати дівчаткам з синдромом Марфана естрогени у віці 7-8 років, щоб викликати передчасне закриття епіфізів і зменшити кінцевий ріст хворих. Лікування дозволило ефективно зменшити ріст, але ці дівчатка потребують спостереження в плані можливого формування сколіозу. Зі зменшенням кінцевого росту може знизитись необхідний серцевий викид і, відповідно, ризик розвитку аневризми аорти.
Естрогени можуть збільшити небезпеку судинних ускладнень. Це необхідно врахувати при призначенні жінкам з синдромом Марфана екзогенних естрогенів, в тому числі і з метою контрацепції.
Профілактика синдрому Марфана шляхом пренатальної діагностики в даний час не здійснюється, тому що невідоме біохімічне порушення, що лежить в основі синдрому. У випадку хворого батька можна ставити питання про штучне запліднення.
Дивіться також:
- Синдром Марфана
- Синдром Марфана (коротка інформація)
- Синдром Марфана (для батьків)
- Синдром Марфана (діаграми розвитку дітей)
Псевдоксантома еластична
Псевдоксантома еластична (ПКЕ) – це спадкове захворювання сполучної тканини, при якому вражаються очі, шкіра, шлунково-кишковий тракт і периферичні судини. Дефект успадковується аутосомно-рецесивно. В деяких родинах передається за аутосомно-домінантним типом.
Перші клінічні прояви з’являються в другому чи третьому десятилітті життя і потім прогресують. На дряблій і нееластичній шкірі видні характерні жовті ксантомоподібні папули, що надають шкірі вигляд “гусячої шкіри”. Різко виражені складки на шкірі, носо-губні, пахвинні, на бічних поверхнях шкіри, пахвинних і пахових складках, животі, стегнах, промежині, інколи бувають гіперкератичні вузли і кальцифікати в дермі. В ІІ і ІІІ десятиріччі життя звичайно з’являються ангіоїдні полоси на сітківці. Внаслідок розривів в мембрані Брука можна знайти крововиливи і хоріоретинальні рубці, які призводять до зниження гостроти зору і сліпоти. Судинні зміни характеризуються оклюзією артерій і кальцифікацією периферичних судин, що і лежить в основі зникнення пульсу на периферичних артеріях, гіпертензії, перемежаючої кульгавості і стенокардії. Часті шлунково-кишкові кровотечі, інколи буває і гематурія.
Про особливі ускладнення при вагітності не повідомлялось. Інколи відмічались післяпологові маткові кровотечі.
Псевдоксантома еластична – це патологія еластичних волокон, але в чому полягає біохімічний дефект невідомо. Пренатальна діагностика неможлива.
Cutix Laxa
Характерне для цієї патології надмірне розтягнення шкіри, що призводить до звисання у вигляді складок по всьому тілу, особливо біля обличчя і очей, що створює вигляд мішкуватих щік і “винуватого обличчя”. Шкіра нееластична, вона не розправляється після зібрання в складку. Можуть уражатись легені (емфізема, кистоз), шлунково-кишковий тракт (дивертикули) і артерії. Типи успадкування – аутосомно-домінантний, аутосомно-рецесивний, рецесивний зчеплений з Х-хромосомою.
Важкі легеневі ускладнення, що властиві рецесивній формі, не дозволяють ураженим дітям дожити до дітородного віку.
Рецесивна, зчеплена з Х-хромосомою, форма зумовлена, ймовірно, дефіцитом ферменту лізолоксидази. Діагноз цієї форми Cutix Laxa можна поставити пренатально, і шляхом визначення статі плода (можуть бути уражені лише хлопчики).
Недосконалий остеогенез
Для недосконалого остеогенезу (НО) характерні остеопороз і переломи довгих трубчастих кісток, що виникають при незначній травмі. Відмічається також прогресуюча деформація довгих кісток, сколіоз, блакитні склери, низькорослість, порушення росту, глухота.
- Тип перший НО – характеризується домінантним успадкуванням і блакитними склерами;
- Тип другий НО – летальний, недосконалий остеогенез новонароджених з “м’ятими кістками”. Він успадковується аутосомно-рецесивно;
- При третьому типі НО склери нормальні, спостерігається прогресуюча деформація кісток. Він успадковується аутосомно-рецесивно;
- У хворих з четвертим типом НО склери білі чи нормальні, важкість переломів варіює. Успадкування аутосомно-домінантне.
Так як у більшості випадків вагітніють жінки з домінантно успадкованим НО, ймовірність народження такими жінками ураженої дитини складає 50%. Домінантно успадкований НО можна діагностувати пренатально. Розродження пропонують проводити шляхом кесарського розтину, враховуючи підвищену небезпеку переломів під час пологів.
Дивіться також:
Артрогрипоз
Артрогрипоз – гетерогенна група порушень, при яких хворі народжуються з недостатністю рухів у багатьох суглобах.
Пологи можуть ускладнюватись тим, що плід не приймає нормальну позицію. Часті сідничне передлежання і поперечне положення плода. У багатьох випадках проводять кесарський розтин у зв’язку з слабістю пологової діяльності чи патологічним положенням плоду. Внаслідок неможливості згинання суглобів у вражених дітей можуть виникати переломи під час пологів. У випадку сімейного артрогрипозу діагноз можна встановити пренатально за допомогою оцінки рухів плода за допомогою УЗД. Відомо, що в більшості випадків жінки народжують нормальних дітей.
Дивіться також:
Література:
- Наследственные синдромы и медико-генетическое консультирование. Атлас-справочник /С.И. Козлова, Н.С. Демикова, Е. Семанова, О.Е. Блинникова. – М.: Практика, 1996.- C. 328-330, 233-234, 147-148, 117, 201-202, 35.
- Шульман Д.Д., Симпсон Д.Л. Наследственные болезни при беременности /Пер. с англ.- М.: Медицина, 1985.- C. 75-86.
- Jones KL. Smith Recognizable Patterns of Human Malformation.- Fifth edition. W. B. Saunders Company. 1997:482-483, 486.
- Warkany J. Congenital Malformations, Notes and Comments. Year Book Medical Publishers, Chicago. 1971:1186-1189, 1011-1015, 824-834, 850-856.
Переглянуто редакційною колегією I.B.I.S.: 09/04/2004
Вплив відеодисплеїв на вагітність
(Video Display and Pregnancy)
Генрих Євгенович Шимонович
Спеціаліст з інформаційного забезпечення
Українсько-Американської Програми запобігання вродженим вадам розвитку
В кінці 70-х перші відеодисплеї (VDT) невпізнано змінили робоче місце американця. Сьогодні 50 мільйонів американських робітників використовують комп’ютери. Багато з них є жінками дітородного віку, яким потрібно знати, чи є відеодисплеї безпечними під час вагітності, чи вони підвищують ризик виникнення вроджених вад розвитку, невиношування вагітності або інших репродуктивних проблем.
Радіація і вроджені вади
На початку 80-х уряд розповсюдив інформацію про те, що відеодисплеї не випромінюють високих доз радіації, що як відомо (в великих дозах) викликає вроджені вади. Відеодисплеї випромінюють рентгенівське проміння, однак воно “розкладається” всередині дисплея.
Ці дослідження були проведені, коли аборти і вроджені вади розвитку дітей трапились у жінок, які використовували відеодисплеї під час вагітності. Ходили чутки про те, що відеодисплеї випромінюють рівень радіації, що перевищує допустимий рівень, як і деякі кольорові телевізори, випущені до 1970 року.
Інші дослідження спростовували ризик радіації для плода. В працівників зустрічались різні вроджені вади, а отже повної достовірності не було. Також важливим був той факт, що вроджені вади не відносились до тих, що викликаються високим рівнем радіації.
Суперечка продовжується
Відеодисплеї випромінюють іншу форму енергії, яку називають електромагнітним полем. Електромагнітні поля також утворюються лініями електропередач, домашньою електропроводкою та електричним обладнанням. На відміну від рентгенівських променів, електромагнітне поле не руйнує клітини і не приносить шкоди генам, і тому вважається безпечним.
В даний час вчені звертають більшу увагу на можливі наслідки для здоров’я через електромагнітне поле, оскільки дослідження у Денвері показали, що діти, які живуть біля електричних ліній високої напруги мали в 1,5-2 рази вищий ризик захворювання раком, особливо лейкемією, ніж інші діти. Комісія Конгресу США по технологіям повідомила, що низькочастотні магнітні поля можуть входити в контакт з окремими клітинами створюючи певні біологічні зміни. Однак значення цих змін невідоме – можливо, що вплив електромагнітного поля створює ризик для вагітних жінок, їхніх дітей, або взагалі для будь-кого.
Деякі вчені вважають думку про те, що вплив електромагнітного поля створює загрозу, сильно перебільшеною, інші вважають, що потенційним впливом на здоров’я не можна нехтувати, поки ми не вивчили в достатній мірі як ця форма енергії може вплинути на організм.
Відеомонітори та аборти
У 1991 році національний інститут безпеки праці та здоров’я видав працю, що вважається найбільш повною та детальною в цьому питанні – в якій говориться про те, що жінки, які працюють з відеодисплеями повний день не мають більшого ризику мати аборт, ніж ті, хто має таку саму роботу і не використовує відеодисплеї. Більшість досліджень можливого ризику відеодисплеїв при вагітності дають такі самі результати.
Це урядове дослідження допомагає спростувати стурбованість вчених з Медичної Програми Кайзер-Перманент Північної Каліфорнії, що говорили у 1988 році про подвоєний ризик абортів серед жінок, що працювали в канцеляріях та користувались відеодисплеями 20 або більше годин на тиждень. Але збільшення кількості абортів серед жінок з професіоналів, що використовували відеодисплеї ту саму кількість годин не було. Різні результати у професіоналів та канцелярських робітників говорять про те, що самі відеодисплеї не є причиною збільшення кількості абортів.
В урядовому дослідженні також говорилось про те, що вагітні жінки, що користувались відеодисплеями не мали вищий ризик ніж ті, що користувались електрообладнанням вдома. Хоча додаткові дослідження по використанню відеодисплеїв під час вагітності все ще проводяться, сучасні дані говорять про те, що відеодисплеї є безпечними для використання під час вагітності.
Інші скарги на стан здоров’я
Багато людей, що працюють з відеодисплеями скаржаться на болі в шиї, спині, зап’ястях, руках та плечах – які лікарі часто розцінюють як болі, викликані постійним м’язевим напруженням – так само, як і напруженням очей. Часто зустрічаються також психологічні стреси; а в деяких дослідженнях говориться про те, що деякі види стресів можуть несприятливо вплинути на вагітність.
Контроль за впливом відеодисплеїв
Вагітні жінки, які не скаржаться на вплив відеомоніторів можуть зменшити їхній вплив, сидячи на відстані витягнутої руки від комп’ютерного дисплею. Потужність електромагнітного поля падає одразу за 24 дюймами. Ні свинцеві екрани, ні будь-який інший захист від радіації не зупиняє електромагнітне поле.
Багато інших фізичних і психологічних стресів від роботи з відеодисплеями можуть пом’якшитись завдяки добре спланованим робочим перервам та зручним дизайном робочого місця.
Оператори відеодисплеїв часто проводять багато часу, не встаючи з робочого місця. Це може призвести до напруження м’язів і поганої циркуляції крові – від чого вагітна жінка може почуватись дискомфортно. Постійне напруження зап’ясть може стати результатом запалення сухожиль, що в свою чергу викликає подразнення нервів, що може проявлятися заціпенінням та болями.
Спеціальні клавіатури, столи та крісла можуть запобігти дискомфорту в області шиї, спини та рук. Крісло повинне мати опору для нижньої частини спини. Дисплей комп’ютера потрібно розмістити на рівні очей або трохи нижче для того, щоб зменшити напруження спини та плечей.
Світло від дисплеїв і постійне зосередження погляду викликає напруження очей. Екрани, зроблені з нерефлекторного скла, з регуляторами яскравості та контрасту, встановлення непрямого освітлення може допомогти зменшити їх напруження.
Рекомендується робити п’ятнадцятихвилинні перерви на вправи кожної години чи дві. Активна прогулянка або потягування зменшує стомленість та збільшує продуктивність. Між перервами можуть допомогти непомітні вправи за комп’ютерним терміналом: пожимання плечима, повороти голови та згинання ніг допомагають циркуляції крові, відпочинку м’язів.
Переглянуто редакційною колегією I.B.I.S.: 05/02/2002
Синдром Меккеля
(Meckel Syndrome)
Сергій Федорович Лапченко
Спеціаліст з інформаційного забезпечення
Українсько-Американської Програми запобігання вродженим вадам розвитку
Синдром Меккеля – це тяжке спадкове захворювання, яке спричиняє народження мертвої дитини, або її смерть в перші дні життя.
Які основні прояви цієї хвороби?
Діти з хворобою Меккеля мають множинні вади розвитку, з яких найтяжчими є ураження головного мозку (зменшений об’єм черепа і мозку та черепно-мозкова кила – випинання мозку через отвір у кістках черепа), кистозні зміни нирок (численні порожнини в тканині нирок, заповнені рідиною), кистозні зміни печінки, вади серця. Крім того, у цих дітей є порушення будови тіла: наявність зайвого пальця на руках і ногах, розщелина губи і піднебіння, недорозвинені статеві органи, великий живіт.
Що є причиною виникнення синдрому Меккеля?
Хвороба виникає у тому випадку, коли дитина отримує у спадок хворобливий ген від кожного з батьків. Така передача генів хвороби називається аутосомно-рецесивним типом успадкування. Це може бути перший випадок захворювання в сім’ї, але слід пам’ятати, що, на жаль, ризик народження кожної наступної дитини з цією хворобою дуже високий і становить 25%.
Чи можна запобігти народженню хворої дитини?
Оскільки методів ефективного лікування цієї хвороби немає, єдиним виходом для сімей, в яких був випадок народження хворої дитини, є медико-генетичне консультування до зачаття та пренатальне (допологове) виявлення ураженого плода.
Переглянуто редакційною колегією I.B.I.S.: 05/02/2002
Дивіться також:
|
|
|
|
|
|





