
Serhiy
Мертвонародження
(Stillbirth)
Г.Є. Шимонович
Спеціаліст з інформаційного забезпечення
Волинського обласного дитячого територіального медичного об’єднання
Коли батьки чують жахливу новину про те, що їхня дитина померла, їхньому горю не має меж. За дуже короткий час їхня радість від народження дитини переходить в сильний біль, викликаний її смертю. Коли смерть плода наступає після 20-го тижня вагітності, це називається мертвонародженням. Ці трагічні смерті трапляються один раз на двісті народжень.
Для більшості батьків ця втрата є цілком несподіваною, тому що більше половини всіх мертвонароджень трапляються в жінок, які нібито не мали проблем. Хоча 14 відсотків мертвонароджень трапляються під час пологів, 86 відсотків трапляються перед їхнім початком. Вагітна жінка може запідозрити, що щось не так з її дитиною, коли вона раптово перестає рухатись.
Як діагностують смерть плода?
Ультразвукове дослідження може підтвердити, що плід помер. Воно також може іноді виявити причину цієї смерті. Після того, як лікар повідомив цю сумну новину батькам, проводиться аналіз крові вагітної жінки, який також може пояснити причину смерті.
Як лікують вагітну жінку?
Якщо медичних показів для термінового переривання вагітності немає, подружня пара може вирішити самостійно, коли вона хоче провести народження мертвої дитини. В більшості випадків, в жінки почнуться пологи на протязі двох тижнів після смерті плода. В цілому, ризик для здоров’я дуже малий, якщо подружня пара вирішить чекати на початок пологів. Однак, оскільки існує емоційна травма при носінні мертвої дитини, більшість пар вирішують зробити примусові пологи. Якщо шийка матки ще не почала розширюватись, жінці дають вагінальні свічки для того, щоб підготувати шийку матки, потім гормон окситоцин (який дають внутрішньовенно), що стимулює родову діяльність. Кесарський розтин рекомендується в тих випадках, коли в жінки виникають проблеми з пологами. Якщо родова діяльність не почалася самостійно на протязі двох тижнів, лікарі рекомендують штучно викликані пологи, тому що існує ризик виникнення небезпечних проблем із згортанням крові.
Що є причиною мертвонародження?
Після народження плода, плід і плацента ретельно обстежуються для того, щоб визначити причину смерті. Часто рекомендують робити аутопсію. Однак навіть після вичерпних аналізів в більше ніж в 1/3 випадків причину смерті неможливо визначити.
Найбільш відомими поширеними причинами мертвонароджень є:
Плацентарні проблеми. Відшарування плаценти – стан, при якому плацента відділяється, частково чи повністю, від стінки матки перед пологами, найчастіше виникає на 35 тиждень вагітності. Результатом може бути важка кровотеча, що загрожує життю матері і дитини, і є причиною важкого кисневого голодування плода, часом закінчуючись його загибеллю. Відшарування плаценти можна діагностувати ультразвуковим дослідженням. Жінки, що палять сигарети, мають на 50 відсотків більший ризик передчасного відшарування плаценти, ніж ті, хто не палить, а ті, що вживають кокаїн під час вагітності подвоюють ризик принаймні вдвічі. Жінки, у яких з’явилась гіпертонія під час вагітності (прееклампсія) також мають подвійний ризик переривання вагітності. Інші проблеми з плацентою, що заважають плоду отримувати достатню кількість кисню та поживних речовин, також можуть спричинити його загибель.
Вроджені вади. Від 5 до 10 відсотків мертвонароджених дітей мають аномалії хромосом, маленьких голкоподібних структур, що містяться в кожній клітині і несуть наші гени. Хромосомні аномалії є найчастішою причиною аборту, що відбувається після 20 тижнів вагітності, і вони також можуть викликати смерть плода в будь-який момент під час вагітності. Інші мертвонароджені діти мають вроджені вади, що не спричинені хромосомними аномаліями, але можуть бути викликані генетичними, природними, або взагалі невідомими факторами.
Затримка зросту. Плоди, які відстають в зрості, можуть померти від асфіксії (брак кисню) і до, і після пологів, і з невідомих причин. Жінки з високим кров’яним тиском мають підвищений ризик народити дитину з відставанням зросту. Ультразвукове дослідження під час вагітності може показати, що плід росте дуже погано, що дає підставу слідкувати за вагітністю дуже ретельно.
Внутрішньоутробні бактеріальні та вірусні інфекції є важливою причиною смерті плода, що може відбутись між 24 та 27 тижнями гестації. Ці інфекції часто не викликають ніяких симптомів у вагітних жінок, що можуть не проходити обстеження до того часу, поки вони не почнуть викликати серйозних ускладнень, такі як смерть плода або передчасне народження (до 37 тижнів вагітності). Після народження, обстеження плаценти та аналізи крові жінки можуть показати, чи власне інфекція викликала смерть плода.
Деякий час вважалось, що брак кисню (асфіксія) під час дуже важких пологів викликала більшість випадків мертвонароджень. Хоча асфіксія під час вагітності іноді все ще викликає смерть плода, вона не є основною причиною. Інші рідкісні причини мертвонароджень включають: переривання пуповини, травми, материнський діабет і високий кров’яний тиск, переношена вагітність (вагітність, що тривала довше, ніж 42 тижні).
Вивчення причин мертвонароджень може допомогти подружжю в його ситуації. Коли причина мертвонародження відома, лікарі можуть дати пораду подружжю щодо ризику мертвонародження, що може відбутись знову під час наступної вагітності. Для більшості подружніх пар ризик буде невеликим. Наприклад, хромосомні аномалії або розрив пуповини мають невеликий шанс виникнути при наступній вагітності. Однак, якщо причиною мертвонародження є хронічне захворювання у матері (таке, як туберкульоз шкіри або високий кров’яний тиск) або якщо воно викликане генетичним захворюванням, то подружжя може мати більший ризик. У таких випадках, рекомендується генетична консультація. Генетик може сказати подружжю про ризик мертвонародження або інші наслідки, що можуть статись при другій вагітності.
Чи можна запобігти мертвонародженню?
Протягом останніх двадцяти років, рівень мертвонароджень знизився приблизно на 50 відсотків. Все це завдяки кращому лікуванню певних захворювань, таких, як підвищений кров’яний тиск у матері та діабет, що може підвищити ризик мертвонародження. Резус конфлікт (несумісність між кров’ю матері і дитини), що до 60-х років був важливою причиною мертвонароджень, якому тепер в більшості випадків можна запобігти (жінкам вводять Rh-негативний імуноглобулін на 28 тиждень вагітності та після народження Rh-позитивної дитини).
Жінок з підвищеним ризиком протягом вагітності ретельно обстежують. Контролюють серцебиття плоду, прискорення або сповільнення якого є ознакою кисневої недостатності. Своєчасна діагностика дає змогу лікарю обрати правильну тактику ведення вагітності та пологів. Сьогодні жінки, в яких діабет і підвищений кров’яний тиск контролюється, мають невисокий ризик мертвонародження.
Лікарі часто вважають, що вагітні жінки з підвищеним ризиком або без нього повинні рахувати рухи плода починаючи з 26 тижнів вагітності. Якщо жінка нараховує менше ніж 10 рухів в день, або якщо вона відчуває, що дитина рухається менше, ніж звичайно, її лікар може порадити зробити аналізи стану плода (наприклад, серцебиття та ультразвук). Якщо аналізи показують, що плід знаходиться під загрозою, існують засоби, що можуть запобігти смерті плода.
Жінки не повинні палити, вживати алкоголь, або використовувати наркотики, що можуть підвищити ризик мертвонародження та інші ускладнення вагітності.
Жінки повинні відразу доповідати їхньому лікарю про будь-які вагінальні кровотечі. Вагінальні кровотечі в другій половині вагітності можуть були ознакою передчасного відшарування плаценти. В цьому випадку терміновий кесарський розтин може врятувати життя дитині.
Жінки, які під час попередньої вагітності мали мертвонародження повинні ретельно обстежуватись при будь-яких ознаках проблем у плода, для того, щоб запобігти ще одному мертвонародженню.
Як можуть батьки справитись із своїм горем?
Подружжя, яке мало мертвонародження, потребує часу на те, щоб пережити своє горе. Оскільки батьки формують зв’язок з своєю дитиною задовго до її народження, природньо, що вони відчувають велику втрату, коли їхня ненароджена дитина помирає. Коли вони переживають свою втрату, вони можуть мати заціпеніння або самозречення, глибоке горе, злість та депресію. Жінка і її партнер можуть справитись із своїм горем по- різному, часом створюючи напруження між собою в той момент, коли вони найбільше потребують одне одного. Можна попросити свого лікаря дати направлення до іншого спеціаліста, який має досвід роботи з подружжям, що втратило дитину. Деяким подружнім парам може піти на користь стати членом групи батьків, що також втратили дітей. В такій групі вони зможуть поділитись своїми почуттями з іншими людьми, що дійсно розуміють, через що вони пройшли, і це може допомогти їм почуватись менш самотніми.
Переглянуто редакційною колегією I.B.I.S.: 15/02/2002
Синдром Мельніка-Нідлса
(Melnick-Needles Syndrome)
О.П. Семененко
Завідувач
Черкаським обласним медико-генетичним центром
Синоніми:
- Мельніка-Нідлса остеодисплазія;
- Melnick-Needles osteodysplasty;
- MNS.
Синдром Мельніка-Нідлса є найбільш важким проявом спектру ото-палато-дигітальних аномалій, що включає також ото-палато-дігітальний синдром тип І, ІІ, і фронто-метафізарну дисплазію.
Частота:
В літературі описано близько 100 випадків.
Основні діагностичні критерії:
- Особливості фенотипу.
- Характерні зміни з боку кісткової системи.
Обличчя:
- Високий лоб, оволосіння чола.
- Екзофтальм, гіпертелоризм.
- Маленьке підборіддя, ретрогенія.
- Кругле обличчя.
- Повні обвислі щоки.
Скелет:
- Нанізм.
- Гіперостоз черепа, затримка закриття тім’ячок.
- Аплазія, гіпоплазія фронтальної, гайморової пазух.
- Нерівні контури діафізів.
- Розширення метафізів.
- Нерівні контури ребер, тонкі ниткоподібні ребра.
- Деформація, нерівний контур ключиці.
- Розширення медіальної частини ключиць; аплазія, остеоліз ключиць.
- Аплазія, гіпоплазія лопаток.
- Вузька грудна клітина.
- Кіфоз, сколіоз.
- Укорочення передньо-заднього розміру хребців.
- Укорочення, викривлення кінцівок.
- Укорочення дистальних фаланг.
- Неосифіковані лонні кістки.
- Коротка клубова кістка, вузьке крило клубової кістки.
- Вальгусне викривлення шийки стегна.
- S-подібна гомілка.
Розвиток:
- Затримка моторного розвитку.
- Збережений інтелект.
Поєднані симптоми:
- Порушення зубного ряду, олігодонтія, затримка прорізання зубів.
- Розщілина піднебіння, язичка.
- Гідронефроз, стеноз уретри.
- Пролапс мітрального, тристулкового клапанів.
- Глухота, приглухуватість.
Лікування: симптоматичне.
Профілактика:
Пошук характерних для синдрому вад скелету при проведенні ультразвукового дослідження під час вагітності.
Етіологія:
Захворювання обумовлено мутацією гена FLNA(300017). Ген картований на хромосомі Хq28. Ген кодує синтез білка filamin. Дослідники припускають, що мутація в гені призводить до порушення регуляції білком filamin процесу розвитку скелету, але невідома генотип-фенотип кореляція.
Чоловіки мають більш важкі клінічні ознаки синдрому, ніж жінки, і майже у всіх випадках помирають до або відразу після народження.
Номер з каталогу МІМ:
309350 Melnick-Needles Syndrome; MNS.
Література:
- Белозеров Ю.М. Детская кардиология. Наследственные синдромы.- 2008, c. 200-201.
- Donnenfeld AE, Conard KA, Roberts NS, et al. Melnick-Needles syndrome іn males: a letal multiple congenital anomalies syndrome. Am J Med Genet 1987 May; 27(1):159-73.
- Eggli K, Giudici M, Ramer J, Easterbrook J, Madewell J. Melnick-Needles syndrome. Four new cases. Pediatr Radiol 1992;22(4):257-61.
- Haar B, Hamel B, Hendriks J, De-Jager J. Melnick-Needles syndrome: indication for an autosomal recessive form. Am J Med Genet 1982 Dec;13(4):469-77.
- Kothari V, Anand R, Chandra J, Garg DP. Melnick Needles syndrome. Indian Pediatr 1995 Apr;32(4):471-5.
- Kristiansen M, Knudsen GP, Soyland A, Westvik J, Orstavik KH. Phenotypic variation in Melnick-Needles syndrome is not reflected in X inactivation patterns from blood or buccal smear. Am J Med Genet 2002 Mar 1;108(2):120-7.
- LaMontagne AE. Urological manifestations of the Melnick-Needles syndrome: a case report and review of the literature. J Urol 1991 May;145(5):1020-1.
- Leyla Akin, Erdal Adal, Mustafa Ali Akin, Selim Kurtoglu. Melnick?Needles Syndrome Associated with Growth Hormone Deficiency: A Case Report. J Clin Res Pediatr Endocrinol 2009 September;1(5):248–251.
- Van der Lely H, Robben SG, Meradji M, Derksen-Lubsen G. Melnick-Needles syndrome (osteodysplasty) in an older male–report of a case and a review of the literature. Br J Radiol 1991 Sep;64(765):852-4.
Переглянуто редакційною колегією I.B.I.S.: 21/09/2012
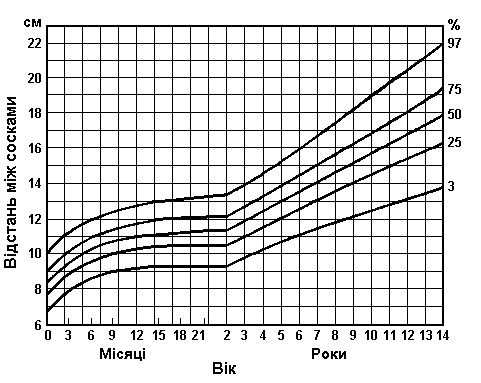
Вимірювання відстані між сосками – співвідношення верхнього та нижнього сегментів тіла
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”

Дані базуються на популяційному дослідженні 2403 дітей від народження до 14 років. 56% з них були чоловічої статі, 44% – жіночої; 2006 – білих, 206 – темношкірих, 43 – східного походження. Feingold M, and Bossert WH: Birth Defects: Orig Art Series X(13), 1974.
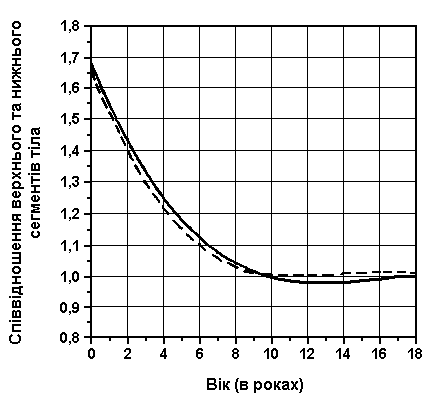
Діаграма співвідношення верхнього та нижнього сегментів тіла: чоловіки показані суцільною лінією, жінки – пунктирною. Engelback [1932], In Wilkins L: Diagnosis and Treatment of Endocrine Disorders in Childhood and Adolescence, Charles C. Thomas, 1965.
Обвід грудей – вимірювання відстані між сосками
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”
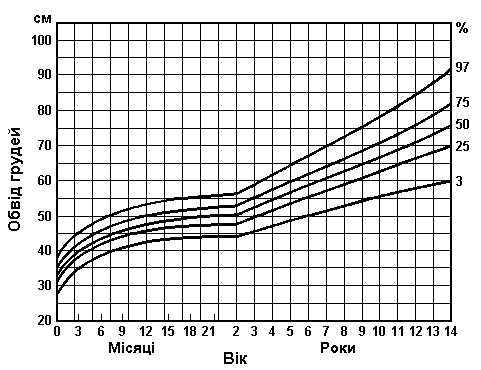
Дані базуються на популяційному дослідженні 2403 дітей від народження до 14 років. 56% з них були чоловічої статі, 44% – жіночої; 2006 – білих, 206 – темношкірих, 43 – східного походження. Feingold M, and Bossert WH: Birth Defects: Orig Art Series X(13), 197.
Дані базуються на популяційному дослідженні 2403 дітей від народження до 14 років. 56% з них були чоловічої статі, 44% – жіночої; 2006 – білих, 206 – темношкірих, 43 – східного походження. Feingold M, and Bossert WH: Birth Defects: Orig Art Series X(13), 1974.
Синдром Маршалла-Сміта
(Marshall-Smith Syndrome)
О.П. Семененко
Лікар-генетик
Черкаського обласного перинатального центру
Синоніми:
- MSS.
Основні діагностичні критерії:
- Прискорений ріст.
- Мілкі орбіти.
- Широкі середні фаланги пальців.
Клінічна симптоматика:
- Затримка моторного розвитку та гіпотонія в ранньому віці.
- Інтелектуальна недостатність (середній показник IQ ~ 50).
Зріст:
Прискорений ріст і значно прискорений кістковий вік. Причина випереджаючого кісткового розвитку невідома, починається внутрішньоутробно. Кістковий вік у деяких пацієнтів в віці двох тижнів відповідав 3-4 рокам. Спостерігається невідповідність зросту вазі.
Краніофаціальні аномалії:
- Довгий череп з випуклим чолом.
- Мілкі орбіти з виступаючими очима.
- Блакитні склери.
- Кирпатий ніс.
- Низька носова перетинка.
Кінцівки:
Широкі проксимальні та середні фаланги, вузькі дистальні фаланги пальців.
Інші аномалії:
- Гіпертрихоз.
- Пупкова кила.
Рідкісні аномалії:
- Атрезія хоан або стеноз, інколи їх поєднання.
- Ларингомаляція.
- Рудиментарний надгортанник.
- Відсутність зубної емалі.
- Глухота та дисплазія вушної мушлі.
- Аномалії мозку включають мікрогірію, церебральну атрофію, агенезію мозолистого тіла.
- Нестабільність краніо-цервікального зчленування.
- Коротка грудина.
- Сколіоз.
- Омфалоцеле.
- Широкий проміжок між І та ІІ пальцями ніг.
- Імунний дефіцит.
Прогноз:
Пацієнти мають постійні дихальні розлади, що проявляються стридором, перерозгинанням шиї та западінням язика. Більшість пацієнтів помирають до 20 місяців життя від пневмонії, аспірації, ателектазів легень, легеневої гіпертензії. Є дані про пацієнтів 7 і 8-річного віку.
Етіологія:
Невідома. Всі випадки спорадичні.
Номер з каталогу МІМ:
602535 Marshall-Smith Syndrome.
Список літератури:
- Adam MP, Hennekam RC, Keppen LD, Bull MJ, Clericuzio CL, Burke LW, Guttmacher AE, Ormond KE, Hoyme HE. Marshall-Smith Syndrome: Natural history and evidence of an osteochondrodysplasia with connective tissue abnormalities. Am J Med Genet 2005;137A:117–124.
- Gorlin RJ, Cohen MM, Hennekam RCM. Syndromes of the Head and Neck. 4-th edition. Oxford University Press. 2001:415 (T11-6).
- Jones KL. Smith’s Recognizable Patterns of Human Malformation. 5th ed. Philadelphia: W. B. Saunders Company 1997:162-163.
- Williams DK, Carlton DR, Green SH, Pearman K, Cole TRP. Marshall-Smith syndrome: the expanding phenotype. J Med Genet 1997;34:842-845.
Переглянуто редакційною колегією I.B.I.S.: 02/04/2008
Ріст та розвиток зубів
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”

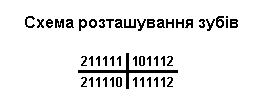
Молочні зуби позначені 1; постійні зуби – 2; відсутні зуби – 0. Верхній ряд показує зуби верхньої щелепи; нижній ряд – нижньої щелепи. Вертикальна лінія означає середню лінію тіла.
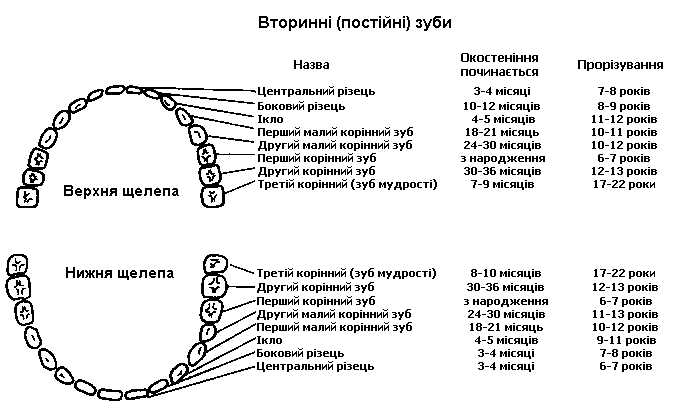
Simon FA, Stevenson RE: Pediatric Patient Care, University of Texas Press, 1975.
Синдром Маршалла
(Marshall Syndrome)
І.В. Красномовець
Лікар-генетик Рівненського обласного клінічного
лікувально-діагностичного центру ім. В. Поліщука
Включення:
Маршалла синдром, Стіклера-Маршалла-Вайсенберга-Вагнера синдром.
Основні діагностичні критерії:
- Гіпоплазія середньої третини обличчя;
- Міопія високого ступеню, що може призвести до відшарування сітківки;
- Катаракта;
- Пласке, широке перенісся;
- Нанізм;
- Глухота по нейросенсорному типу, що призводить до затримки мовного розвитку;
- Гіпоплазія/ дисплазія епіфізів;
- Розширення метафізів.
Допоміжні симптоми:
- Екзофтальм, косоокість;
- Випуклий лоб/скронева ділянка;
- Пігментний ретиніт/дегенерація сітківки;
- Маленький ніс;
- Короткий ніс;
- Кирпатість;
- Довгий фільтр;
- Відкритий рот;
- Розщілина піднебіння/язичка;
- Мікрогенія;
- Платиспондилія;
- Вкорочення кінцівок;
- Гіпереластичність суглобів;
- Артрит/ поліартрит/артроз.
Супутні ознаки:
- Затримка розумового розвитку в спорадичних випадках;
- Макроцефалія/мегаленцефалія;
- Епікант;
- Гіпертелоризм;
- Глаукома;
- Гіпоплазія нижньої щелепи;
- Астигматизм;
- Розщілина верхньої губи латеральна;
- Тонкі губи;
- Нерівний, подвійний ряд зубів;
- Коротка грудна клітка/тіло;
- Сутулість/кіфоз;
- Сколіоз;
- Лордоз;
- Контрактури суглобів/артрогрипоз;
- Камподактилія кисті.
Дані обстежень:
- Відшарування/випадання скловидного тіла;
- Пігментний ретиніт/дегенерація сітківки;
- Гіпоплазія/атрофія сітківки/аномалія Рігера;
- Вивих/дистопія/ектопія кришталика;
- Ретролентальна мембрана/фіброплазія;
- Персистування a. hyaloid/первинного скловидного тіла;
- Помутніння/дисплазія скловидного тіла;
- Хореоретинальна атрофія;
- Атрофія/гіпоплазія зорового нерва;
- Гіпопігментація очного дна.
Рентгенодіагностика:
- Розширення кісток черепа;
- Гіпертрофія/деформація суглобів;
- Вальгусне викривлення шийки стегна;
- Широка коротка шийка стегна;
- Гіпоплазія/дисплазія голівки стегна;
- Внутрішньочерепні кальцифікати;
- Аплазія/гіпоплазія гайморової пазухи;
- Гіпоплазія фронтальних синусів.
Лабораторні дані: наявність мутантного гена COL11A1 (120280.0002).
Етіологія: аутосомно-домінантний тип успадкування.
Частота: 1 на 60000.
Співвідношення статей: Ч1 : Ж1.
Патогенез:
Вивчений недостатньо. В основі – мутація COL11A1 гена (120280.0002).
Вік прояву:
Перші підозри можуть з’явитися в неонатальному періоді внаслідок високого рівня стигматизації. Вже на першому році життя може спостерігатись міопія. У віці старше 10-ти років з’являється катаракта, що спонтанно розсмоктується у дорослих. У ранньому віці діагностують зниження слуху і затримку розвитку мови при переважно нормальному інтелекті.
Історія хвороби/прогноз:
Пренатальний/неонатальний період. Обстеження жінки в пренатальному періоді, яка є лише носієм мутантного гена COL11A1 не дає можливості запідозрити у плода синдром Маршала, тобто характерні ознаки та симптоми їй непритаманні.
При першому неонатальному огляді може звернути на себе увагу високий рівень стигматизації дитини: типовим є обличчя хворих – гіпертелоризм, маленький, короткий ніс, запале перенісся, відкритий рот, опуклий виступаючий лоб, екзофтальм, гіпоплазія середньої третини обличчя. Дещо рідше можна виявити розщілину піднебіння/язичка, розщілину верхньої губи збоку, гіпоплазію нижньої щелепи, коротку грудну клітку. На першому році життя до множинних дизморфій приєднується міопія. У віці старше 10-ти років з’являється катаракта, що спонтанно розсмоктується у дорослих. У зв’язку з високим ступенем міопії може розвинутись відшарування сітківки.
Дитинство. Виявляється зниження слуху за нейросенсорним типом, що може прогресувати до повної глухоти, і, як правило, призводить до затримки розвитку мови. Інтелектуальний розвиток нормальний.
Дорослий вік. У спорадичних випадках зустрічалась розумова відсталість. Характерним є нанізм. Спонтанно розсмоктуються сформовані в шкільному віці катаракти. У дорослому віці можна простежити характерні для синдрому зміни кісткової, суглобної систем у вигляді гіпоплазії/дисплазії епіфізів, розширення метафізів, вкорочення кінцівок, платиспондилії, гіпереластичності суглобів, артритів, поліартритів, артрозів, контрактур суглобів, внутрішньочерепних кальцифікатів, їх виявляють при радіологічних обстеженнях, комп’ютерній томографії та ін.
Відносно фертильності, відомості, що свідчать про порушення у хворих репродуктивної здатності, відсутні.
Ускладнення:
Основними з них є відшарування сітківки внаслідок важкої міопії. В результаті поступового зниження гостроти слуху може розвинутись повна нейросенсорного типу глухота, що в свою чергу призводить до глибокого порушення розвитку мови. Дана симптоматика перебігає “під маскою” розумової відсталості, що затруднює встановлення вірного діагнозу. Деформація і аномалії розвитку хребців та кісток призводять до розвитку остеопорозу.
Диференційний діагноз: акродизостоз, синдром Стіклера, синдром Робінова.
Лікування:
Симптоматичне. Оперативні втручання використовують для корекції офтальмологічної патології.
Запобігання:
- Первинне/вторинне генетичне консультування;
- Рання діагностика;
- Раннє профілактичне лікування.
Номер з каталогу МІМ:
154780 Marshall Syndrome.
Література:
- Козлова С.И., Демикова Н.С., Семанова Е., Блинникова О.Е. Наследственные синдромы и медико–генетическое консультирование.- М.: Практика, 1996.- C. 149-150.
- Конигсмарк Б.В., Горлин Р.Д. Генетические и метаболические нарушения слуха.- М.: Медицина, 1980.- C. 123-126.
- Jones K.L. Smith’s Recognizable Patterns of Human Malformation. W.B.Saunders Company, Philadelphia 1997:252-254.
- Marshall Syndrome. OMIM #154780.
Переглянуто редакційною колегією I.B.I.S.: 11/11/2002
Дивіться також:
Синдром МакК’юна-Олбрайта
(McCune-Albright Syndrome)
Наталія Олегівна Зимак-Закутня
Завідуюча Хмельницькою
медико-генетичною консультацією
Синоніми: поліостотична фіброзна дисплазія.
Вперше захворювання описане незалежно МакК’юном (McCune) і Брач (Bruch) у 1937 році та Олбрайтом (Fuller Albright, 1937, 1938). Пізніші дослідження розширили його клінічний спектр: окрім кісткових, шкірних та проявів з боку яйників, описані ураження інших ендокринних залоз, а також серця, печінки. Lichtenstein (1938) запропонував назву “поліостотична фіброзна дисплазія”, підкреслюючи характер скелетних уражень при синдромі.
Основні діагностичні критерії:
- фіброзна дисплазія;
- шкірна гіперпігментація;
- автономна ендокринна гіперфункція;
- передчасне статеве дозрівання.
Етіологія та патогенез:
Синдром МакК’юна-Олбрайта виникає внаслідок постзиготичної соматичної мутації у гені GNAS1 (OMIM #139320 Gnas Complex Locus; GNAS), що кодує альфа-субодиницю стимулючого G-білка (Gsa). Останній, з’єднуючи рецептори поверхні клітин з інтрацелюлярними білками, активізує або інактивує сигнальні каскади. Стимулючий G-протеїн активується у момент зв’язування гормону чи іншого ліганду з клітинними рецепторами. Активована Gsa-субодиниця, відщеплюючись від рецептора, натомість зв’язується з аделатциклазою, стимулючи підвищення рівня внутрішньоклітинного циклічного аденозин-монофосфату (цАМФ). Gsa-субодиниця інактивується, повторно зв’язується з рецептором і стає знову доступною для гормонально опосередкованої реактивації.
Специфічна мутація, що зумовлює розвиток синдрому МакК’юна-Олбрайта, зачіпає сайт, відповідальний за інактивацію Gsa-субодиниці, внаслідок чого одного разу активована альфа-субодиниця G-протеїну залишається активною протягом тривалого часу, незважаючи на відсутність прямої гормональної стимуляції рецепторів, що, відповідно, приводить до постійно підвищеного рівня внутрішньоклітинного цАМФ. А це, у свою чергу, посилює мітоз та стимулює обмінні процеси у клітині.
Специфічний фенотип формуватиметься залежно від типу клітин та тканин, що містять мутацію:
- тривале підвищення активності Gsa-субодиниці у меланоцитах зумовлює появу характерної гіперпігментації у вигляді плям кольору кави з молоком;
- постійна стимуляція пострецепторного сигнального каскаду цАМФ у фолікулах яйників приводить до формування кіст, продукування естрогенів та формування гонадотропін-незалежного передчасного статевого дозрівання.
Подібний механізм (підвищення рівня інтрацелюлярного цАМФ) пояснює усі інші ендокринні та неендокринні прояви синдрому МакК’юна-Олбрайта.
Чим раніше в ембріогенезі виникла мутація, тим більша кількість тканин буде уражена, оскільки кожна дочірня клітина мутантної нестиме таку ж зміну. Відповідно, при більш пізньому виникненні мутації формуються локальні, обмежені ураження з неповним (2 чи 3 класичні прояви) та м’якшим перебігом захворювання. Результатом мутації, котра виникає після завершення диференціації на специфічні клітинні лінії, буде розвиток аденом (подібні мутації виявлені у соматотрофних аденомах гіпофіза, ізольованих гіперфункціонуючих вузлах щитовидної залози).
Gsa-мутація у гермінативних клітинах летальна, саме тому не спостерігається аутосомно-домінантна передача захворювання.
Діагностика:
Діагностика синдрому МакК’юна-Олбрайта базується на наявності щонайменше двох фенотипових проявів, пов’язаних з активацією Gsa-мутації.
- Передчасне статеве дозрівання, зумовлене надлишком естрогенів: ріст молочних залоз, менструальні кровотечі (іноді на 1-му році життя), розвиток статевого оволосіння.
- Фіброзна дисплазія із широким спектром фенотипових проявів – від незначних, із асимптоматичним перебігом, до виражених деформацій трубчастих кісток, хребта, черепа. Останні особливо проблематичні, оскільки можуть приводити до втрати зору, слуху. Кісткові ураження мають тенденцію до одностороннього розташування.
- Гіперпігментація шкіри типу “плям кольору кави з молоком” – неправильних контурів пігментовані макули (від світло- до темно-коричневого кольору), що мають схильність до сегментарної, переважно однобічної локалізації. Обриси “плям кольору кави з молоком” неправильні, часом їх порівнюють з контуром узбережжя Майн, на відміну від гіперпігментацій при нейрофіброматозі, котрі, здебільшого, менших розмірів та правильніших контурів (порівнюють із контуром узбережжя Каліфорнії).
- Гіпертиреоз як прояв гіперметаболічного стану: тахікардія, гіпертензія, тремор, безсоння, схуднення.
- Виражена гіпофосфатемія (нечасто) з проявами рахіту: викривлення ніг, рахітичні зміни ребер, низький зріст.
- Синдром Кушинга розвивається також відносно рідко: затримка росту, зниження м’язового тонусу, кушингоїдність обличчя, артеріальна гіпертензія.
- Гіперпродукція гормону росту аденомою гіпофіза також приводить до розвитку гіпертензії, зниження м’язового тонусу, проте на фоні значного збільшення ростових параметрів (аж до гігантизму або акромегалії у старшому віці).
Лабораторні обстеження:
- Визначення рівня естрадіолу, ФСГ, ЛГ у сироватці крові: підвищення естрадіолу на фоні зниженого рівня гонадотропних гормонів гіпофіза свідчить на користь гонадотропін-незалежного передчасного статевого розвитку, властивого синдрому МакК’юна-Олбрайта. Оскільки, секреція естрогенів має пульсуючий характер, це іноді вимагає повторних обстежень для виявлення цих відхилень. Віддиференціювати передчасне статеве дозрівання центрального генезу допомагає ЛГ-рілізінгфактор-стимулюючий тест (пригнічення або різке зниження рівня ФСГ, ЛГ після введення фактрілу характерне для синдрому МакК’юна-Олбрайта).
- Підвищення рівня сироваткового кортизолу при зниженні АКТГ. Для диференціювання АКТГ-залежної та АКТГ-незалежної гіперпродукції кортизолу рекомендується проведення дексаметазонового тесту.
Тип успадкування: спорадичні випадки.
Популяційна частота:
Точні дані відсутні. Синдром МакК’юна-Олбрайта – відносно рідкісне, спорадичне генетичне захворювання.
Співвідношення між статтю:
Гонадотропін-незалежне передчасне статеве дозрівання набагато частіше спостерігається в осіб жіночої статі. Інші ж клінічні прояви синдрому зустрічаються з однаковою частотою у осіб обох статей.
Вік маніфестації:
У важких випадках, що супроводжуються множинними ендокринними ураженнями, діагностика можлива уже незадовго після народження. Повідомлялось про гіпертиреоз, синдром Кушинга у неонатальному періоді. Фіброзна дисплазія, пігментація, гіпофосфатемія також, зазвичай, розвиваються у ранньому дитячому віці. Передчасне статеве дозрівання описане навіть у 4-місячної дівчинки, проте здебільшого це трапляється після 1 року життя.
Гонадотропін-продукуючі аденоми гіпофіза та функціональні тиреоїдні аденоми, зумовлені Gsa-мутацією, можуть виявлятись у будь-якому віці.
Генетичний ризик: для сибсів та потомства уражених осіб невисокий.
Диференційна діагностика:
- гігантизм та акромегалія;
- Синдром Кушинга та побічна дія терапії глюкокортикоїдами;
- гіпертиреоз;
- гіпофосфатемічний рахіт;
- нейрофіброматоз;
- передчасне статеве дозрівання центрального генезу.
Лікування та нагляд:
Передчасне статеве дозрівання: агоністи гонадотропін-рілізинггормону неефективні, враховуючи його гонадотропін-незалежний характер. Основним препаратом є тестолактон – інгібітор ароматази (блокує перетворення тестостерону у естрадіол, таким чином знижуючи рівень циркулюючих естрогенів). Хірургічного втручання зрідка потребують кісти яйників великих розмірів, однак, зрозуміло, що це є виключно паліативним заходом, оскільки при існуючій гіперестрогенії зберігається причина рецидивування кіст. Отож, хірургічне лікування не рекомендується для лікування передчасного статевого дозрівання при синдромі МакК’юна-Олбрайта.
Необхідний ретельний нагляд дитячого ендокринолога з тривалим моніторингом зросту та розвитку: підвищений рівень естрогенів сприяє передчасному закриттю зон росту, зменшуючи кінцевий ріст пацієнтів. До того ж, рання естрогенна стимуляція може зумовити активізацію гіпоталамічно-гіпофізарно-яйникової вісі з розвитком гонадотропін-залежного передчасного статевого дозрівання.
Фіброзна дисплазія: на жаль, ефективні методи медикаментозної терапії відсутні. Обнадійливими видаються останні повідомлення про застосування біфосфонатів, особливо щодо зменшення больових проявів та запобігання прогресуванню кісткових уражень. У випадку виникнення переломів часто необхідне хірургічне втручання. Пацієнти потребують нагляду дитячого ортопеда. Прогресуючі деформації, виражений больовий синдром можуть свідчити про патологічні переломи і вимагати відповідного ортопедичного втручання. При ураженні кісток черепа необхідний регулярний контроль гостроти та полів зору, а також сурдологічне обстеження.
Гіпертиреоз: особливого нагляду потребують діти, що перенесли тиреоїдектомію чи лікування радіоактивним йодом. Оскільки тиреоїдні гормони життєво важливі для нормального розвитку та функціонування мозку, такі пацієнти повинні проходити обстеження ендокринолога із визначенням гормонів щитовидної залози щоквартально, і лише після 3 років припустимо зменшити частоту відвідувань до 1 разу на 4-6 місяців.
Гіпофосфатемічний рахіт: лікування вираженої гіпофосфатемії аналогічне лікуванню Х-зчепленого гіпофосфатемічного рахіту – додавання фосфатів (1,5-2,5 г/добу у 5 прийомів) разом із кальцитріолом (0,25–0,5 мг/добу).
Інфантильний синдром Кушинга: у зв’язку з відсутністю ефективних медикаментозних методів лікування АКТГ-незалежного синдрому Кушинга, єдиною рекомендацією залишається двобічна адреналектомія із подальшою замісною терапією глюкокортикоїдами та мінералокортикоїдами.
Гігантизм/акромегалія: найефективнішим є лікування гонадотропінпродукуючих аденом. Варто пам’ятати, що променева терапія може спричиняти виникнення саркоматозних змін в оточуючих фіброматозно змінених кістках черепа. Застосовується також (із змінним успіхом) октреотид – аналог соматостатину тривалої дії. До хірургічного лікування вдаються лише у випадку ураження аденомою зорових трактів.
Пренатальна діагностика: специфічна не розроблена.
Прогноз: життя при вчасній діагностиці та адекватному лікуванні сприятливий.
Профілактика: профілактичні заходи передбачають переважно попередження можливих ускладнень.
Номер з каталогу МІМ:
174800 Mccune-Albright Syndrome; MAS.
Література:
- Abs, R.; Beckers, A.; Van de Vyver, F. L.; De Schepper, A.; Stevenaert, A.; Hennen, G.: Acromegaly, multinodular goiter and silent polyostotic fibrous dysplasia: a variant of the McCune-Albright syndrome. J. Endocr. Invest. 13: 671-675, 1990.
- Albright, F.; Butler, A. M.; Hampton, A. O.; Smith, P.: Syndrome characterized by osteitis fibrosa disseminata, areas of pigmentation and endocrine dysfunction, with precocious puberty in females: report of five cases. New Eng. J. Med. 216: 727-746, 1937.
- Albright, F.; Scoville, B.; Sulkowitch, H. W.: Syndrome characterized by osteitis fibrosa disseminata, areas of pigmentation, and a gonadal dysfunction: further observations including the report of two more cases. Endocrinology 22: 411-421, 1938.
- Axelrod, L.: Bones, stones and hormones: the contributions of Fuller Albright. New Eng. J. Med. 283: 964-970, 1970.
- Bianco, P.; Kuznetsov, S. A.; Riminucci, M.; Fisher, L. W.; Spiegel, A. M.; Robey, P. G.: Reproduction of human fibrous dysplasia of bone in immunocompromised mice by transplanted mosaics of normal and Gs-alpha-mutated skeletal progenitor cells. J. Clin. Invest. 101: 1737-1744, 1998.
- Chanson, P.; Dib, A.; Visot, A.; Derome, P. J.: McCune-Albright syndrome and acromegaly: clinical studies and responses to treatment in five cases. Europ. J. Endocr. 131: 229-234, 1994.
- Comite, F.; Shawker, T. H.; Pescovitz, O. H.; Loriaux, D. L.; Cutler, G. B., Jr.: Cyclical ovarian function resistant to treatment with an analogue of luteinizing hormone releasing hormone in McCune-Albright syndrome. New Eng. J. Med. 311: 1032-1036, 1984.
- Feuillan, P. P.; Foster, C. M.; Pescovitz, O. H.; Hench, K. D.; Shawker, T.; Dwyer, A.; Malley, J. D.; Barnes, K.; Loriaux, D. L.; Cutler, G. B., Jr.: Treatment of precocious puberty in the McCune-Albright syndrome with the aromatase inhibitor testolactone. New Eng. J. Med. 315: 1115-1119, 1986.
- Feuillan, P. P.; Jones, J.; Oerter, K. E.; Manasco, P. K.; Cutler, G. B., Jr.: Luteinizing hormone-releasing hormone (LHRH)-independent precocious puberty unresponsive to LHRH agonist therapy in two girls lacking features of the McCune-Albright syndrome. J. Clin. Endocr. Metab. 73: 1370-1373, 1991.
- Giovannelli, G.; Bernasconi, S.; Banchini, G.: McCune-Albright syndrome in a male child: a clinical and endocrinologic enigma. J. Pediat. 92: 220-226, 1978.
- Hall, R.; Warrick, C.: Hypersecretion of hypothalamic releasing hormones: a possible explanation of the endocrine manifestations of polyostotic fibrous dysplasia (Albright’s syndrome). Lancet I: 1313-1316, 1972.
- Jones K.L. Smith’s recognizable patterns of human malformation. W.B. Saunders company. 1988:372–374.
- Lichtenstein, L.: Polyostotic fibrous dysplasia. Arch. Surg. 36: 874-898, 1938.
- McCune, D. J.; Bruch, H.: Progress in pediatrics: osteodystrophia fibrosa. Am. J. Dis. Child. 54: 806-848, 1937.
- Nager, G. T.; Holliday, M. J.: Fibrous dysplasia of the temporal bone: update with case reports. Ann. Otol. Rhinol. Laryng. 93: 630-633, 1984.
- Olsen, B. R.: A rare disorder, yes; an unimportant one, never. (Editorial) J. Clin. Invest. 101: 1545-1546, 1998.
- Reitzik, M.; Lownie, J. F.: Familial polyostotic fibrous dysplasia. Oral Surg. 40: 769-774, 1975.
- Schwindinger, W. F.; Francomano, C. A.; Levine, M. A.: Identification of a mutation in the gene encoding the alpha subunit of the stimulatory G-protein of adenylyl cyclase in McCune-Albright syndrome. Proc. Nat. Acad. Sci. 89: 5152-5156, 1992.
- Syndromes of the head and neck (fourth edition) by Robert J. Gorlin, M.Michael Cohen, Raol C. M. Hennekam, Oxford University Press. 2001:334-337.
- Weinstein, L. S.; Shenker, A.; Gejman, P. V.; Merino, M. J.; Friedman, E.; Spiegel, A. M.: Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. New Eng. J. Med. 325: 1688-1695, 1991.
- Yoshimoto, M.; Nakayama, M.; Baba, T.; Uehara, Y.; Niikawa, N.; Ito, M.; Tsuji, Y.: A case of neonatal McCune-Albright syndrome with Cushing syndrome and hyperthyroidism. Acta Paediat. Scand. 80: 984-987, 1991.
Переглянуто редакційною колегією I.B.I.S.: 27/08/2003
Ширина вуха – вимірювання фільтру
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”

Середнє значення ширини вуха (в мм)
1 2 3 4 5 6 7-8 9-10 11-12 13-14 15-16 17-18 18-20 20-30 30-40 40-50 50-60 60-70 70-80 |
31 32 32 32 33 33 34 35 35 35 36 36 36 36 36 36 36 36 36 |
28 31 31 32 32 33 33 33 33 33 33 33 33 33 33 33 33 33 34 |


Goodman RM, Gorlin RJ: The Malformed Infant and Child, Oxford University Press, 1983. (Дані для нижнього графіку взяті з Feingold M, Bossert WH: Birth Defects: Orig Art Series X(13), 1974.).
Вимірювання довжини вуха
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”

Дані обстеження 2403 (56% хлопчиків, 44% дівчаток; 2006 білих, 206 темношкірих, 43 монголоїдної раси) дітей, від народження до 14-річного віку. За Feinhold M, Bossert WH: Birth Defects: Orig Art Series X(13), 1974.


Середня довжина вуха – хлопчики (суцільна лінія), дівчатка (пунктирна лінія). Goodman RM, Gorlin RJ: The Malformed Infant and Child, Oxford University Press, 1983.
|
|
|
|
|
|





