
Uncategorized
Гіпоспадія
(Hypospadias)
Ж.І. Мартинюк
Лікар-неонатолог
Волинського обласного дитячого територіального медичного об’єднання
Основні діагностичні критерії:
Гіпоспадія – вроджене недорозвиття сечопускового каналу, пов’язане з порушенням внутрішньоутробного розвитку уретральної трубки, з характерною варіабельністю локалізації зовнішнього отвору сечопускового каналу, ступеня недорозвитку його по довжині, наявності та вираженості вад розвитку інших відділів сечостатевої системи.
Частота:
Виявляють у одного з 300-400 новонароджених хлопчиків, у одної з 20-25000 новонароджених дівчаток.
Етіологія:
В більшості випадків невідома. Полігенний тип успадкування доведений. В деяких сім’ях гіпоспадія передається від батька до сина, як аутосомно-домінанта з статевою обумовленністю.
Патогенез:
На третьому тижні розвитку мезенхімні клітини, які походять з ділянки первинної смужки, мігрують довкола клоакальної мембрани, формуючи клоакальні складки, які в краніальному напрямку сполучаються, утворюючи статевий горбок. Каудально вони поділяються на сечівникові складки спереду і відхідникові – ззаду. З обох боків сечівникових складок утворюється пара виростів – статеві валики, які формують калиткові валики у зародків чоловічої статі та великі соромітні губи у зародків жіночої статі.
Розвиток зовнішніх статевих органів у чоловіків перебуває під впливом андрогенів, продукованих яєчками плода, і характеризується швидким видовженням статевого горбка, який називається прутнем. Видовжуючись, прутень підтягує сечівникові складки вперед, формуючи латеральні стінки сечівникової борозни, вона простягається вздовж каудального боку видовженого прутня, але не досягає головки. Епітеліальне вистелення борозни має ендодермальне походження і утворює сечівникову пластинку. Наприкінці третього місяця сечівникові складки змикаються над сечівниковою пластинкою, утворюючи сечівник прутня, який не досягає верхівки прутня. Найдистальніша частина сечівника формується протягом четвертого місяця, коли ектодермальні клітини верхівки головки прутня проникають усередину і утворюють короткий епітеліальний тяж, згодом в ньому виникає просвіт – це зовнішне вічко сечівника. Якщо сечівникові складки зрощуються не цілковито, то аномальні отвори сечівника містяться на нижній поверхні прутня. В рідкісних випадках зовнішній отвір сечівника пролягає вздовж калиткового шва.
Клініка:
Різноманітна, залежить від форми гіпоспадії, вираженості порушення сечопуску, віку. При гіпоспадії сеча виділяється через аномальний зовнішній отвір сечопускового каналу.


Перша ступінь. Гіпоспадія головки або вінцева, зовнішній отвір сечопускового каналу розміщений в ділянці прикріплення вуздечки крайньої плоті, головка дещо опущена, покрита надмірною крайньою плоттю тільки зверху. Звуження струменю сечі, незвичайна форма статевого члена виявляються з народження.
Друга ступінь. Стовбурова гіпоспадія, зовнішній отвір сечопускового каналу розміщений на різному рівні по середній лінії вентральної поверхні статевого члена проксимальніше головки, статевий член зігнутий донизу. Сечопуск можливий тільки в положенні сидячи.
Третя ступінь. Калиткова гіпоспадія, калитка розщеплена по середній лінії, зовнішній отвір сечопускового каналу розташований в жолобі між половинками калитки, статевий член недорозвинутий, вигнутий донизу, має форму крючка. Сечопуск по жіночому типу, при затримці яєчок в пахових каналах роздвоєна калитка схожа на великі статеві губи, а зігнутий пеніс на збільшений клітор.
Четверта ступінь. Промежинна гіпоспадія, зовнішній отвір сечопускового каналу розміщений на промежині, недорозвиток статевих органів поєднується з крипторхізмом.
Гіпоспадія у дівчаток розпізнається не завжди, зовнішній отвір сечопускового каналу локалізується на передній стінці піхви, безпосередньо за входом або на деякій віддалі – це часткова гіпоспадія; повна гіпоспадія – коли зовнішній отвір сечопускового каналу знаходиться на рівні входу в сечовий міхур.
Поєднані симптоми:
Аномалії нирок і сечовивідних шляхів: підковоподібна нирка, ектопія нирки, подвоєння уретри. Вроджені вади серця, глухота, атрезія прямої кишки, гідроцефалія, омфалоцелє, Vater асоціація.
Диференціальний діагноз:
Синдром нечутливості до андрогенів частковий; брахіоскелетогенітальний синдром; викривлення прутня; дистопія зовнішнього отвору сечопускового каналу; G синдром; істинний гермафродитизм; гіпертелоризму-гіпоспадії синдром; окулоцереброфаціальний синдром, тип Кауфмана; пеноскротальна транспозиція; Сміта-Лемлі-Опіца синдром; вроджена фістула уретри; хромосоми 4p синдром; мозаіцизм 45х/46XY; недостатність 3-бета-гідроксістероїд дегідрогенази; недостатність 5-альфа-редуктази; недостатність 17-альфа-гідроксилази; недостатність 17-20-десмолази; дефект 20-22-десмолази.
Ускладнення:
Хорда – 5%, стеноз отвору уретри – 8%, мікропеніс, розщеплення калитки, крипторхізм – 8-15%, гідроцелє – 16%, вроджена пахова кила – 8%.
Вік маніфістації: з народження.
Генне картування і групи зчеплення генів: невідомі.
Співвідношення статей: Ч10 : Ж1.
Ризик для сибсів пробанда: 11%.
Ризик для батьків пробанда: у 8% випадків знаходять гіпоспадію у батька пробанда.
Лікування:
При звуженні зовнішнього отвору сечопускового каналу меатотомію проводять в перші місяці життя дитини; випрямлення статевого члена проводиться до трьох років життя, з обов’язковим завершенням пластики сечопускового каналу до 6-7 років, щоб в школу дитина пішла здоровою.
Номер з каталогу МІМ:
146450 Hypospadias, Autosomal
Література:
- Aarkodg D. Current concepts: maternal progestins as a possible cause of hypospadias. New Engl J Med 1979;300:75-79.
- Bauer S, et al. Hypospadias. Urol Clin North Am 1981; 8:559-571.
- Frudman M, et al. Uncomplicated familial hypospadias: evidence for autosomal recessive inheritance. Am J Med Genet 1985;21:51-55.
- Svensos J. Male hypospadias: 625 cases, associated malformation and possible etiological factors. Acta Paediats 1979;68:587-592.
- Sweet RA, et al. Study of the incidence of hypospadias in Rochester, MN, 1949-1970, and a case control comparison of possible etiologic factors. Mayo Clin Proc 1974;49:52-58.
- Taur V, et al. Hypospadias in a consanguineous family (letter). Am J Med Genet 1987; 27:487-489.
Переглянуто редакційною колегією I.B.I.S.: 26/11/2002
Гіпертрофічна кардіоміопатія
(Hypertrophic Cardiomyopathy)
О.В. Тростянська
Лікар-генетик
Медико-генетична консультація, м. Ялта
Гіпертрофічна кардіоміопатія (ГКП) – первинне захворювання міокарда, що характеризується дифузною або обмеженою гіпертрофією міокарда шлуночка (як правило, лівого) без розширення або з незначним зменшенням його порожнини.
Поширеність:
Частота виявлення гіпертрофічної кардіоміопатії у дорослого населення коливається в широких межах від 0,17% до 0,5-1,1%. У дитячому віці хворі на гіпертрофічну кардіоміопатію складають від 41% до 58% всіх хворих на кардіоміопатію.
Етіологія:
Захворювання генетично детерміноване, передається за аутосомно- домінантним типом успадкування з високою пенетрантністю та різною експресивністю. Серед хворих переважають особи чоловічої статі – до 70%. Відомо понад 50 мутацій у локусах генів, що контролюють структуру й функції скоротливих білків міокарду. Генні мутації зачіпають найчастіше ген β-важких ланцюгів міозину, ген серцевого тропоніну, ген α-тропоміозину та ген білка С, який зв’язує міозин.
Ген β-важких ланцюгів міозину кодує 30% всіх білків серця. Мутації цього гену виявляють у 30-50% сімейних випадків гіпертрофічної кардіоміопатії. Ген локалізований на 14 хромосомі.
Ген серцевого тропоніну–Т – кодує синтез 5% білків міофібрил. Відомо 8 мутацій цього гену, локалізованого на першій хромосомі. Виявляють у 15% сімей з гіпертрофічною кардіоміопатією. Клінічною особливістю цього типу ГКП є помірна гіпертрофія міокарду лівого шлуночка, але високий ризик раптової смерті.
Ген α-тропоміозину кодує синтез близько 5% білків міофібріл. Ген локалізований на 15 хромосомі. Мутацією цього гена обумовлено 5% випадків сімейної гіпертрофічної кардіоміопатії, яка характеризується частим приєднанням дилатації лівого шлуночка, рефрактерної серцевої недостатності, раптової серцевої смерті.
Ген міозин-зв’язуючого білка С локалізований на 11 хромосомі. Дуже рідко його мутації є причиною гіпертрофічної кардіоміопатії.
Таким чином, гіпертрофічна кардіоміопатія – захворювання генетично гетерогенне. У розвитку гіпертрофічної кардіоміопатії істотну роль відіграють й інші фактори. Ангіотензин–перетворюючий фермент: монозиготи по мутації цього гену мають високий вміст ангіотензину-2, що стимулює гіпертрофію кардіоміоцитів; надмірний вплив катехоламінів, порушення вивільнення й поглинання кальцію (Са) саркоплазматичним ретікулумом з підвищенням внутріклітинної концентрації Са, – фактори, що сприяють гіпертрофії міокарду.
Класифікація:
Існує декілька класифікацій гіпертрофічної кардіоміопатії. Всі вони групуються на клініко-анатомічному принципі.
Табл. 1. Робоча класифікація за О.А. Мутаф’яном:
| 1. Спадковість | 1. Сімейна форма 2. Спорадична форма |
| 2. Характер і локалізація гіпертрофії | 1. Симетрична 2. Асиметрична:
|
| 3. Вираженість і характер обструкції | 1. Необструктивна 2. Обструктивна:
|
| 4. Стадії хвороби й градієнт тиску | 1 стадія – градієнт тиску до 25 мм рт. ст. 2 стадія – градієнт тиску до 35 мм рт. ст. 3 стадія – градієнт тиску до 80 мм рт. ст. |
| 5. Ускладнення | 1. Синкопальний синдром 2. Аритмічний синдром 3. Тромбоемболічний синдром 4. Серцева недостатність 1-2-3-4 |
Патанатомія:
Маса серця збільшена. Виражена гіпертрофія міокарду лівого шлуночку: товщина стінки перевищує 13-15 мм. Гіпертрофія міокарду може бути симетричною й асиметричною. Асиметрична форма зустрічається значно частіше – до 95%. Як правило, виявляється потовщеною верхня частина міжшлуночкової перетинки (60-95%), рідше – верхівка, ще рідше – серединношлуночкова частина. Характерно, що відношення товщини міжшлуночкової перетинки до товщини задньої стінки лівого шлуночку більше 1,3. Кардіоміоцити збільшені, деформовані, хаотично розташовані. Вихровий хід волокон може спостерігатися й при інших захворюваннях серця, але при гіпертрофічній кардіоміопатії вираженість цієї ознаки максимальна. Ядра в кардіоміоцитах збільшені, ексцентрично розташовані, зі світлим “вінцем”. Рясні грубі прожилки сполучної тканини оточують м’язові клітини або їх пучки.
Клініка, діагностика:
Гіпертрофічна кардіоміопатія формується антенатально, але перші ознаки захворювання можуть бути виявлені як у новонародженого, так і в зрілому віці. За даними Оводової Н.Ф. (1984), сімейні форми частіше виявляються до 15 років, а спорадичні – пізніше 25 років.
Багато хворих протягом тривалого часу почувають себе задовільно, ведуть активний спосіб життя, дозволяють собі підвищені фізичні навантаження.
Найбільш характерними для гіпертрофічної кардіоміопатії є скарги на біль в області серця, серцебиття, непритомність, запаморочення, задишку, які виникають після фізичних і психоемоційних навантажень.
Больовий синдром спостерігається у третини хворих, має ниючий, колючий характер у загрудинній ділянці, не зникає від прийому коронаролітиків.
Задишка – одна із ранніх скарг. З’являється спочатку після фізичного навантаження, а потім й у спокої. Синкопальний синдром зустрічається у 10-40% дорослих хворих (Амосова О.Н., 1999), у 26-80% хворих дітей і підлітків (Леонтьєва І.В. 2000). Провокується фізичними й психоемоційними навантаженнями. Частота коливається від поодиноких епізодів протягом життя до щоденних випадків.
Аритмічний синдром спостерігається у 32-45% хворих. Проявляється серцебиттям, відчуттями перебоїв у роботі серця. Частіше виявляють порушення ритму, рідше – провідності. У дітей порушення ритму бувають рідше, ніж у дорослих.
Електрокардіографічні зміни більш ніж у 60% хворих виявляються перш за все ознаками гіпертрофії міокарду лівого шлуночка, гіпертрофії міжшлуночкової перетинки (30-50%). За наявності мітральної регургітації й перевантаження лівого передсердя часто фіксується Pmitrale. Часто спостерігають шлуночкову екстрасистолію, суправентрикулярну тахікардію. Характерним для гіпертрофічної кардіоміопатії є феномен WPW (Вольфа-Паркінсона-Вайта). Прогностично несприятливим, через можливість раптової смерті, є виявлення подовженого інтервалу QT. Багаторічні катамнестичні спостереження за дітьми з порушенням ритму (G. Muller, 1992) при гіпертрофічній кардіоміопатії показали, що у 50% аритмія асоціюється з раптовою смертю. У 1/3 хворих з необструктивною формою гіпертрофічної кардіоміопатії може не бути істотних змін на електрокардіографії.
Систолічний шум може бути виявлений випадково або у зв’язку зі скаргами на біль в серці. Він є раннім симптомом захворювання і, фактично, складається з двох різних шумів: перший на верхівці, тривалий, з проведенням в ліву аксилярну ділянку, другий – мінливого характеру, з відривом від першого тону, краще вислуховується в 3-4 міжребер’ї зліва від грудини. На фонокардіограмі шум систоли має ромбоподібну форму, розташований в першій половині систоли або мезосистоли, не доходить до другого тону.
Одним із важливих діагностичних методів є еходоплерокардіографія. Основні ознаки гіпертрофічної кардіоміопатії при цьому дослідженні:
- потовщення стінки лівого шлуночка більше 13-15 мм;
- збільшення співвідношення товщини міжшлуночкової перетинки до товщини задньої стінки лівого шлуночка більше 1,3-1,5;
- зменшення порожнини лівого шлуночка;
- мітрально-септальний контакт;
- раннє або середньосистолічне прикриття стулок аортального клапану;
- субаортальний градієнт тиску більше 20 мм. рт. ст.
Диференційна діагностика:
Схожість гіпертрофічної кардіоміопатії з рядом вроджених вад серця та інших серцево-судинних захворювань обумовлена загальними клінічними проявами у вигляді больового синдрому, синкопи, систолічним шумом, гіпертрофією лівих відділів серця, порушенням ритму.
- Стеноз устя аорти, клапанний або субаортальний фіброзний, допомагає виявити еходоплерокардіографія: при стенозі устя аорти спостерігається постстенотичне розширення висхідної аорти, градієнт тиску визначається на рівні аортального клапану. При фіброзному субаортальному стенозі немає гіпертрофії міжшлуночкової перегородки, а обструкція носить фіксований характер.
- Дефект міжшлуночкової перетинки. При дефекті міжшлуночкової перетинки часто спостерігається тремтіння систоли, шум вислуховується від народження, носить грубий характер, пансистолічний. На ехокардіографії знаходять дефект міжшлуночкової перетинки, дилатації лівого шлуночка.
- Коарктація аорти – “дорослий тип”. При цій ваді систолічний шум краще вислуховується в міжлопатковій ділянці зліва. Діагностиці допомагає визначення пульсу на руках і ногах: відсутність або різке ослаблення на стегнових артеріях і напружений на радіальній (в області ліктьового згину). На ехокардіографії виявляють обструкцію на рівні перешийка аорти.
- Недостатність мітрального клапана, частіше пов’язана з ревматизмом. Враховують дані анамнезу, лабораторні дослідження. При ехокардіографії – потовщення й деформація порожнини лівого шлуночку.
- Серце спортсмена. В діагностиці допомагає характерний сімейний анамнез, а також ознаки, не характерні для серця спортсмена: зменшення порожнини лівого шлуночка, порушення релаксації лівого шлуночку, нерівномірна гіпертрофія міокарда, порушення ритму й провідності.
Перебіг захворювання:
Перебіг може бути безсимптомним (але з характерними змінами на електрокардіографії, ехокардіографії), маніфестним та маломаніфестним, але з фатальним результатом.
Лікування симптоматичне, паліативне хірургічне – корекція асиметричної гіпертрофії й дефектів мітрального клапана.
Номер з каталогу МІМ:
- 115196 Cardiomyopathy, Familial Hypertrophic, 3; CMH3
- 115197 Cardiomyopathy, Familial Hypertrophic, 4; CMH4
- 188840 Titin; TTN
- 191044 Troponin I, Cardiac; TNNI3
- 608758 Cardiomyopathy, Familial Hypertrophic, 10; CMH10
- 608751 Cardiomyopathy, Familial Hypertrophic, 8; CMH8
- 192600 Cardiomyopathy, Familial Hypertrophic 1; CMH1
Література:
- Беленков Ю.Н., Агеев Ф.Н., Мареев В.Ю. Парадоксы сердечной недостаточности: взгляд на проблему на рубеже века //Сердечная недостаточность.- 2000.– Т.1, №1.– С.4-6.
- Билоконь Н.А., Кубергер М.Б. Болезни сердца и сосудов у детей (руководство для врачей).– Москва, 1987.
- Кардіологія дитячого віку /Ред. П.С. Мощич, В.М. Сидельников, Д.Ю. Крівченя.- Київ, 1986.
- Мутафьян О.А. Кардиомиопатии у детей и подростков.- С.-Петербург, 2003.
- Шевченко Е.Г., Таболин В.А., Котлукова Н.П. и др. Генетические синдромы у детей с ВПС //Вести аритмологии.- 2000.- №18.– С.102–03.
Переглянуто редакційною колегією I.B.I.S.: 23/07/2007
Первинна легенева гіпертензія
(Primary Pulmonary Hypertension)
Н.Г. Тандура
Лікар-неонатолог
Волинського обласного дитячого територіального медичного об’єднання
Мінімальні діагностичні критерії:
- Підвищення тиску в легеневій артерії до 25 мм рт. ст. в покої і 30 мм рт. ст. при фізичному навантаженні;
- Гіпертрофія міокарда правого шлуночка;
- Розширення стовбура легеневої артерії;
- Фіброз і фіброеластоз інтими легеневих артеріол;
- Фібриноїдно-некротичний артеріїт, тромбоз дрібних гілок легеневої артерії.
Частота:
17 випадків на 10000 аутопсій (Pittman D, 1989 р). За даними інших авторів 1-2 випадки на 1000000 населення в рік.
Патогенез:
Виявлення частої моноклональної ендотеліальної клітинної проліферації при первинній легеневій гіпертензії (ПЛГ) означає, що можлива схожість з неопластичними процесами. З часом наступає фіброз артерій, вони втрачають еластичність. Є тенденція до появи згустків крові в них. Відповідно до розвитку ПЛГ серцевий м’яз збільшується, праве передсердя розширюється, поступово слабне і втрачає здатність перекачувати достатньо крові.
Клініка:
Первинна легенева гіпертензія на ранніх стадіях не дає виражених клінічних проявів. Першими скаргами є поява задишки при помірному фізичному навантаженні, болі в серці, серцебиття, слабкість.
Ціаноз розвивається в більш пізні стадії, з’являється тахікардія, епігастральна пульсація, яка пов’язана з вираженою гіпертрофією правого шлуночка, акцент і розщеплення другого тону над легеневою артерією. Інколи у лівого края грудини і в четвертому міжребер’ї вислуховується систолічний шум (відносна недостатність 3-стулкового клапану). Можуть бути вторинні запальні зміни з боку легень. У хворих відмічається потовщення нігтьових фаланг (барабанні палички). В термінальній фазі констатують збільшення і болючість печінки.
Дані обстежень:
При рентгенологічному дослідженні знаходять різке розширення проксимальних відділів легеневої артерії, послаблення легеневого малюнку в периферичних відділах легень, збільшення розмірів правих відділів серця, розширення кореня легень і посилена їх пульсація.
На ЕКГ є ознаки раннього перевантаження правого шлуночка і його гіпертрофія.
Заключний діагноз підтверджується при зондуванні порожнини правого серця і легеневої артерії, при якому встановлюють точні цифри тиску в легеневій артерії.
Генне картування:
Ген первинної легеневої гіпертензії локалізований на хромосомі 2q33.
Етіологія:
Первинна легенева гіпертензія відноситься до захворювань з аутосомно-домінантним типом успадкування, але можлива аутосомно-рецесивна передача з перевагою у жінок. Описані сімейні форми цієї патології. Відмічається гетерогенність патологічних змін навіть при сімейних випадках захворювання. Від 6% до 10% випадків ПЛГ є спадковими. ПЛГ може бути обумовлена вродженим судинним дефектом, при якому зберігається фетальний тип гемодинаміки. Фіброз і фіброеластоз м’язевого шару легеневих артеріол ймовірно пов’язаний з генетично обумовленим дефектом гладких м’язових волокон. Причиною ПЛГ може бути спадковий дефіцит тромбоцитів з високим рівнем 5-гідрокситриптаміну. Є дані, що в основі первинної легеневої гіпертензії лежать нейрогуморальні впливи з ангіоспазмом і наступними морфологічними змінами в судинах. Певне значення мають імунні порушення, інфекції, тромбоз судин, аномалії розгалуження легеневої артерії. Є повідомлення про зв’язок ПЛГ з порушенням метаболізму простагландинів в легенях.
Фактори ризику:
- Прийом медикаментів для схуднення (фенфлюрамін/фентермін);
- Прийом наркотиків;
- Оральні контрацептиви або замісна гормональна терапія;
- Проживання високо в горах;
- Вагітність.
Диференціальний діагноз:
З вторинною легеневою гіпертензією обумовленою захворюванням легень (хронічний бронхіт, емфізема), легеневою тромбоемболією, вродженою і набутою патологією серця і судин, хворобами сполучної тканини.
Співвідношення статей: Ж2 : Ч1.
Вік маніфестації:
Середній вік захворювання – 36 років, хоча воно може проявитись і в дитячому віці.
Перебіг:
Немає співвідношення між часом виникнення, віком і клінічними проявами. В деяких пацієнтів, особливо дітей, хвороба прогресує дуже швидко. Розрізняють 3 форми перебігу ПЛГ:
- швидку;
- помірну;
- повільно-прогресуючу.
Тяжка, швидко прогресуюча ПЛГ, яка протікає з різким ціанозом, тяжкою задишкою, поліцитемією, вираженою гіпертрофією правих відділів серця, нерідко визначається, як синдром Аерза.
Прогноз:
Несприятливий. Смерть наступає при прогресуючій правошлуночковій недостатності.
Лікування:
Позитивний ефект відмічається при застосуванні а-адреноблокаторів (толазолін, фентоламін), антагоністів кальцію. Широко використовуються судиннорозширюючі препарати: еуфілін, нітрогліцерин, пролонговані форми нітратів, антикоагулянти, а також препарати, які сприяють покращенню серцевої діяльності: серцеві глікозиди, протиаритмічні, кортикостероїди, сечогінні, киснева терапія. Легенева трансплантація являється єдиною альтернативою в лікуванні хворих, яким консервативне лікування вже не допомагає. Рівень виживання з легеневою трансплантацією – приблизно 60% за рік і 37% за 5 років. Ускладнення трансплантації – відторгнення тканин та інфекції.
Номер з каталогу МІМ:
- 178600 Pulmonary Hypertension, Primary; PPH1
- 265400 Pulmonary Hypertension, Primary; Autosomal Recessive
Література:
- Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, Venetos G, Kalachikov S, Cayanis E, Fischer SG, Barst RJ, Hodge SE, Knowles JA. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet. 2000 Sep;67(3):737-44.
- Gaine SP, Rubin LJ. Primary pulmonary hypertension. Lancet. 1998 Aug 29;352(9129):719-25.
- Hendrix GH. Familial primary pulmonary hypertension. South Med J. 1974 Aug;67(8):981-3.
Переглянуто редакційною колегією I.B.I.S.: 19/02/2003
Співвідношення обводу голови та гестаційного віку новонародженого (на рівні моря)
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”.
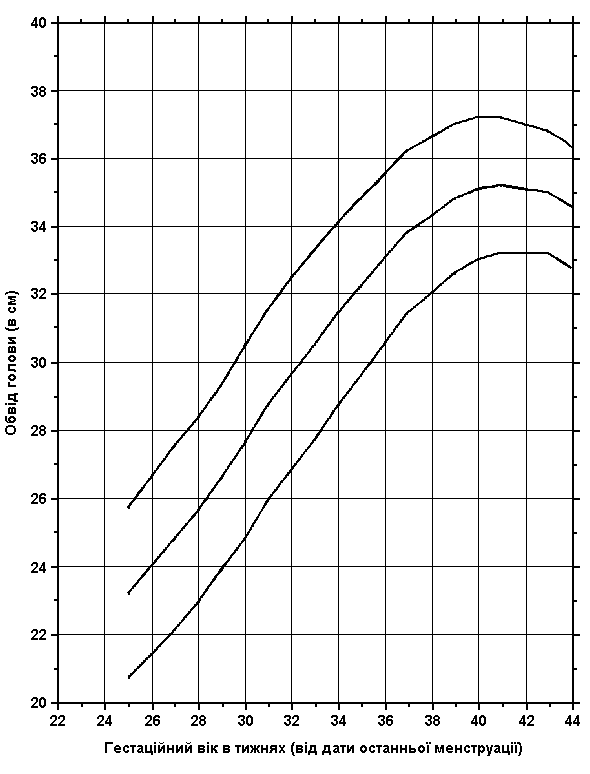
3-й, 50-й та 97-й персентилі обводу голови при народженні співставляються з гестаційним віком. Дані отримані на основі досліджень 300 немовлят (145 хлопчиків та 155 дівчаток), народжених на одній висоті над рівнем моря з Монреалем (Канада) впродовж 1959-1963 років. Враховувались дані на дітей білої раси, з одноплідної вагітності, народжених у віці 25-44 тижнів гестації. Не включались діти з великими вродженими вадами розвитку, діти, народжені від матерів з цукровим діабетом, та з невідомим терміном гестації. Усі вимірювання робились протягом перших 36 годин життя. Usher R, and McLean F: Intrauterine growth of live-born Caucasian infants at sea level: Standards obtained from measurements in 7 dimensions of infants born between 25 and 44 weeks of gestation, J Pediatr 74:901, 1969.
Співвідношення росту та гестаційного віку новонародженого (на рівні моря)
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”.
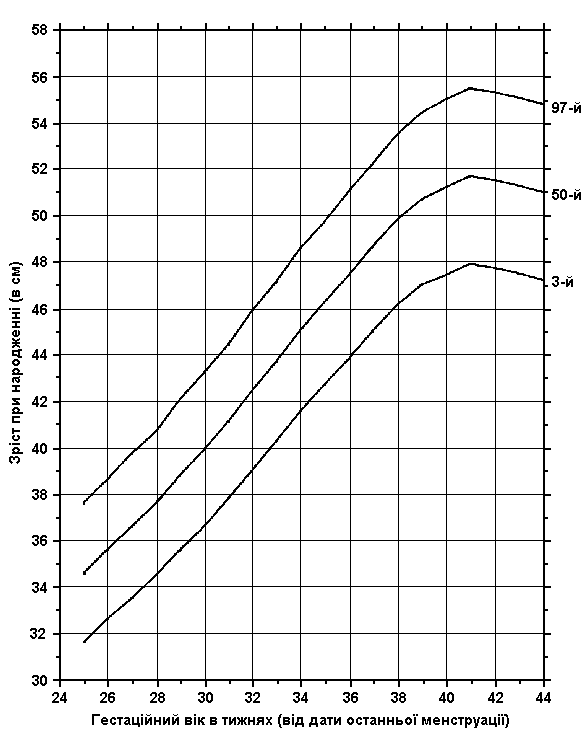
3-й, 50-й та 97-й персентилі зросту при народженні співставляються з гестаційним віком. Дані отримані на основі досліджень 300 немовлят (145 хлопчиків та 155 дівчаток), народжених на одній висоті над рівнем моря з Монреалем (Канада) впродовж 1959-1963 років. Враховувались дані на дітей білої раси, з одноплідної вагітності, народжених у віці 25-44 тижнів гестації. Не включались діти з великими вродженими вадами розвитку, діти, народжені від матерів з цукровим діабетом, та з невідомим терміном гестації. Усі вимірювання робились протягом перших 36 годин життя. Usher R, and McLean F: Intrauterine growth of live-born Caucasian infants at sea level: Standards obtained from measurements in 7 dimensions of infants born between 25 and 44 weeks of gestation, J Pediatr 74:901, 1969.
Співвідношення ваги та гестаційного віку новонародженого (на рівні моря)
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”.

3-й, 50-й та 97-й персентилі ваги при народженні співставляються з гестаційним віком. Дані отримані на основі досліджень 300 немовлят (145 хлопчиків та 155 дівчаток), народжених на одній висоті над рівнем моря з Монреалем (Канада) впродовж 1959-1963 років. Враховувались дані на дітей білої раси, з одноплідної вагітності, народжених у віці 25-44 тижнів гестації. Не включались діти з великими вродженими вадами розвитку, діти, народжені від матерів з цукровим діабетом, та з невідомим терміном гестації. Усі вимірювання робились протягом перших 36 годин життя. Usher R, and McLean F: Intrauterine growth of live-born Caucasian infants at sea level: Standards obtained from measurements in 7 dimensions of infants born between 25 and 44 weeks of gestation, J Pediatr 74:901, 1969.
Фетальний гідантоіновий синдром
(Fetal Hydantoin Syndrome)
Т.В. Віговська
Зав. неонатологічним центром Волинського
обласного дитячого територіального медичного об’єднання
Фенітоін (діфенілгідантоін) – це антиконвульсант, який широко застосовується проти судом всіх типів за виключенням petit mal при епілепсії. Дослідами на тваринах доведений тератогенний ефект похідних гідантоіну у вигляді розщелини піднебіння, мікромелії, аномалії нирок та гідроцефалії.
Основні діагностичні критерії:
Найчастіше зустрічається гіпоплазія нігтів і дистальних фаланг, з затримкою росту та розвитку, розщелина губи та піднебіння, гіпоплазія середньої частини обличчя.
Клінічні прояви:
Найбільш повна клінічна картина включає пре- і постнатальну затримку росту з проявами гіпоплазії нігтів та іноді – дистальних фаланг обох рук і ніг. Більш повна характеристика дисморфічних рис включає: плоске перенісся з помірно вираженим епікантом, помірний гіпертелоризм та страбізм. Рідше зустрічається недорозвинений фільтр, помірна крилоподібна шийна складка, птоз, розщелина губи та піднебіння. Характерна дерматогліфічна картина у вигляді сплощених дуг на подушечках пальців та дігіталізація 1-го пальця руки. Серед дітей з фетальним гідантоіновим синдромом близько 30% пацієнтів мають різного ступеня затримку розвитку та розумову відсталість. Наявність мікроцефалії значно збільшує ризик розвитку розумової відсталості.
Ускладнення:
Відставання у розвитку, помірна до середньовираженої розумова відсталість приблизно у третини уражених осіб.
Поєднані симптоми:
Аномалії серця, центральної нервової системи та сечостатевого тракту.
Патогенез:
Базується на зниженій здатності лейкоцитарної епоксидгідроксилази метаболізувати гідантоін до нетоксичних форм. Оскільки зареєстровані випадки, коли вражається один плід із дизиготної двійні, а здоровий сибс при цьому зберігає високий рівень епоксидгідроксилази, не можна виключити спадковий дефект біохімічних процесів.
Співвідношення статей: Ч1 : Ж1
Частота випадків:
Повний симптомокомплекс фетального гідантоінового синдрому задокументований приблизно у 7-10% дітей, народжених матерями, що отримували цей препарат.
Ризик повторення для сибсів: невідомий.
Ризик повторення для нащадків:
Мінімальний за умови, що мати не буде вживати гідантоін під час вагітності.
Вік маніфестації:
Внутрішньоутробну затримку росту можна діагностувати пренатально, заключний діагноз ставиться при народженні чи в ранньому віці.
Генне картування і групи зчеплення генів: невідомі.
Профілактика:
Уникання гідантоіну під час вагітності.
Лікування:
Хірургічна корекція великих дефектів, спеціальні навчальні і тренувальні програми.
Прогноз:
Грунтується на гостроті уражень.
Список літератури:
- Buehler BA, et al. Prenatal prediction of risk of the fetal hydantoin syndrome. New England Journal of Medicine 1990;322:1567-1572.
- Buehler BA, Hanson JW. Fetal hydantoin syndrome. In: Buyse ML, ed. Birth Defects Encyclopedia. Dover: Center for Birth Defects Information Services, Inc., 1990:714-715.
- Dansky LV, Finnell RH. Parental epilepsy, anticonvulsant drugs, and reproductive outcome: Epidemiologic and experimental findings spanning three decades; 2: Human studies. Reproductive Toxicology 1991;5:301-355.
- Finnell RH, Kansky LV. Parental epilepsy, anticonvulsant drugs, and reproductive outcome: Epidemiologic and experimental findings spanning three decades; 1: Animal studies. Reproductive Toxicology 1991;5:281-299.
- Jones KL. Fetal hydantoin syndrome. In: Smith’s Recognizable Patterns of Human Malformation. 5th ed. Philadelphia: W. B. Saunders Company, 1997:559.
- Kelly TE. Teratogenicity of anticonvulsant drugs. I: Review of the literature. American Journal of Medical Genetics 1984;19:413-434.
Переглянуто редакційною колегією I.B.I.S.: 05/02/2002
Генітальний герпес
(Genital Herpes)
А.С. Вакульчик
Спеціаліст з інформаційного забезпечення
Українсько-Американська Програма запобігання вродженим вадам розвитку
Генітальний герпес – це захворювання, що передається статевим шляхом і, при зараженні вагітної жінки, спричиняє пошкоджуючу дію на організм новонародженої дитини. За даними Центру Контролю за здоров’ям США близько 45 мільйонів американців мають генітальний герпес, при цьому кожного року реєструється коло 500,000 нових випадків, 1,000 з яких складають новонароджені. Більшість жінок, що хворіють на генітальний герпес, мають здорових дітей, але незважаючи на це, певна кількість з них передає вірус герпесу своїм дітям. Таким чином для вагітних жінок особливо важливо виявити симптоми захворювання і вчасно звернутись до лікаря для отримання кваліфікованої допомоги.
Які симптоми генітального герпесу?
Зазвичай, через певний проміжок часу після зараження герпесом, в області геніталій з’являються групи пухирців, що супроводжуються свербінням та болючістю. Після вскриття пухирців, на їх місці залишаються болісні виразки. Часто з’являються інші уретральні чи вагінальні зміни, а також лихоманка, втома, біль. Перший напад, триває, як правило, протягом трьох місяців. При повторних нападах прояви інфекції стають менш вираженими, тривалість їх зменшується. Діагностика герпесу лікарем можлива шляхом виділення вірусної культури.
Однак, у багатьох заражених вірусом герпесу, як перший, так і повторний напади, можуть не проявлятися симптомами і лишаються недіагностованими. В результаті, більшість інфікованих не знають про це. Проте ці асимптомні, чи приховані, інфекції можуть також передаватись іншим, включаючи новонароджених.
Що викликає герпес?
Герпес викликають так звані віруси простого герпесу, що схожі на віруси, що викликають вітряну віспу і оперізуючий лишай.
Існують два основні типи вірусів герпесу: тип 1, що пов’язаний з висипкою навколо рота і на губах; і тип 2, який зазвичай викликає генітальну висипку.
Однак, і інші типи вірусу герпесу можуть уразити оральні і генітальні області. Таким чином, вагітна жінка, хвора на генітальний герпес, викликаний іншою формою герпесу, також може передати інфекцію своїй дитині.
Після інфікування віруси герпесу можуть потрапити всередину нервових клітин, і тоді їх знищення імунною системою організму стає неможливим. Але під впливом певних умов, ці віруси можуть активізуватися та викликати повторні загострення. До цього призводять різноманітні впливи навколишнього середовища – такі як спека, переохолодження; крім того статеві відносини, менструація, лихоманка та емоційний стрес. В середньому людина може мати від 4 до 7 загострень протягом року. З часом частота їх зменшується, а прояви інфекції стають менш серйозними з кожним разом.
Які симптоми інфекції герпесу у новонародженої дитини?
У деяких новонароджених, інфікованих вірусом герпесу, з’являється висипка на шкірі та навколо рота чи спостерігається ураження очей. Коли інфекція обмежується цими органами, більшість дітей в подальшому розвиваються нормально, хоча серйозні пошкодження нервової системи і очей все одно залишаються.
Однак, часто герпетична інфекція у новонароджених поширюється на головний мозок та інші внутрішні органи. Інфікована дитина може бути збудженою, погано їсть і страждає від нападів. Навіть при проведеному лікуванні близько половини дітей з поширеною інфекцією, що уражує внутрішні органи, а також 10% тих, хто має інфекцію головного мозку, помирають. У 50% дітей, що пережили поширену інфекцію, розвиваються незворотні зміни головного мозку, що може спричинити розумову відсталість, дитячий церебральний параліч, сліпоту і глухоту.
Який ризик для плода становить герпес під час вагітності?
Принаймні, одна з п’яти вагітних жінок інфікована вірусом герпесу, хоча більшість про це і не здогадується. На щастя, лише меншість з них може передати інфекцію своїм дітям чи страждати від інших ускладнень вагітності.
Шанс передати інфекцію своїм дітям під час пологів для жінки, що вперше заражена генітальним герпесом безпосередньо перед пологами складає від 30% до 50%, якщо у неї є виражені симптоми захворювання. Ризик настільки високий через те, що організм щойно інфікованої жінки ще не має вироблених антитіл, що можуть допомогти захистити дитину протягом пологів. Дослідження показують, що близько 2% вагітних жінок, які до того не мали герпесу, інфікуються під час вагітності.
Жінки, що були інфіковані раніше і мали явну чи “приховану” інфекцію мають лише 3% шансів інфікування їхніх дітей під час вагінальних пологів. Іноді прихована інфекція може активізуватись протягом вагітності. Такі жінки мають мало шансів на те, що їхні діти будуть інфіковані. На жаль, доступний на даний час аналіз крові не може визначити, стара інфекція чи нова.
Деякі дослідження також показують, що жінки, які були вперше інфіковані герпесом в пізніх термінах вагітності, можуть мати підвищений ризик дострокових пологів чи ризик мати дитину з малою вагою при народженні. Жінки із загостренням старих інфекцій не ризикують стикнутися з такими ускладненнями.
Хоча більшість дітей інфікуються герпесом від своїх матерів під час пологів, у дуже рідких випадках дитина може бути інфікована до народження. Дитина також може інфікуватись герпесом після народження, якщо хтось з оральним герпесом поцілує її. Ви не повинні торкатись чи цілувати дитину після того, як торкались такого інфікованого.
Як лікується інфекція герпесу?
Існує три види антивірусних препаратів (ацикловір, валацикловір, і фамцикловір), які можуть зменшити тривалість нападів і допомогти пом’якшити симптоми. Якщо їх приймати профілактично, то вони зменшують кількість загострень у пацієнтів з частими інфекціями. Ці ліки зазвичай не рекомендуються під час вагітності. Проте, ацикловір (який не асоціюється з вродженими вадами за більш як 10 років використання), часом призначається вагітним жінкам з початковою стадією герпесу і жінкам з серйозними герпесними ускладненнями. Ацикловір випускається як у таблетках, так і у вигляді мазі, але оральні ліки більш ефективні.
Інфікованих новонароджених лікують ацикловіром чи іншим антивірусним препаратом, що називається відарабін. Ці препарати є досить ефективними при лікуванні локальної інфекції з ураженням очей, шкіри чи роту, але вони менш ефективні, якщо інфекція потрапила у головний мозок чи внутрішні органи.
Як жінка може запобігти герпесу під час вагітності?
Для запобігання передачі герпесу чи будь-якої іншої статевої хвороби протягом вагітності, жінка повинна знати, що її партнер не інфікований. Цього можна досягти, якщо подружжя дотримується виключно моногамних стосунків. Якщо ж партнер вагітної жінки мав герпес раніше, йому слід утриматись від статевих стосунків, якщо у нього є симптоми інфекції; і користуватись презервативом, якщо у нього немає симптомів (оскільки, він не може знати, чи є у нього прихована інфекція). Деякі лікарі рекомендують подружжям утримуватись від статевих відносин протягом останнього триместру, якщо чоловік мав генітальний герпес і жінка не була інфікована.
Чи можливо захистити дитину від інфекції, якщо мати інфікована?
При наявності у вагітної жінки під час пологів симптомів, що вказують на активну інфекцію (першу чи повторну), дитину можна вберегти від інфікування шляхом пологів через кесарський розтин.
Для більшості жінок з повторним герпесом, що не мають симптомів, безпечними є вагінальні пологи. Однак, дослідження показують, що 75% жінок, що народили дітей, інфікованих вірусом герпесу, не мали симптомів на момент пологів. Кращого захисту дітей від матерів, що мають приховану інфекцію герпесу, на даний час немає.
До недавнього часу, деякі лікарі рекомендували жінкам, що вже мали герпес, пройти тест на виділення вірусу в терміні вагітності від 34 тижнів, що могло б допомогти виявити асимптомні інфекції.
На жаль, ці тести не можна використати протягом пологів, оскільки визначення результату триває від одного до трьох днів. На сьогодняшній день опрацьовуються нові тести, які б зробили можливими більш швидку діагностику, визначення прихованої інфекції протягом пологів, та запобігання інфекції новонароджених.
Переглянуто редакційною колегією I.B.I.S.: 15/02/2002
Новонароджена дитина з невизначеними (двозначними) геніталіями
(Intersex – Hermaphroditism)
Ю.С. Коржинський
Доктор медичних наук, професор
Завідувач кафедри педіатрії і неонатології
Львівського державного медичного університету
Народження дитини з невизначеними зовнішніми статевими органами (геніталіями) є однією із складних медичних та соціальних проблем, що ставить ряд складних завдань перед лікарями та болюче сприймається батьками. Статева невизначеність новонародженого немовляти нерідко перетворюється в соціальну кризу. Це вимагає швидкого і в той же час докладного та поглибленого обстеження немовляти перед тим, як визначиться стать дитини та розпочнеться лікування. Обстеження такої дитини і визначення її статі вимагає участі лікарів різних спеціальностей та використання ряду додаткових методів діагностики. Воно повинно бути проведено так швидко, як це можливо, але, в той же час, зайняти стільки часу, скільки потрібно. У більшості випадків після народження такої дитини виникає потреба переведення її в спеціалізоване відділення, де є найкращі умови для проведення такого обстеження.
В цілому ряді випадків визначення статі новонародженої дитини з невизначеними геніталіями може бути дуже складним завданням, і до обговорення при прийнятті остаточного рішення варто залучати батьків дитини. Кожен такий випадок вимагає індивідуального підходу. Рекомендується не реєструвати народження дитини і не давати дитині ім’я, якщо можливо, доки не визначено стать виховання. Необхідно усвідомлювати, що в окремих випадках, незважаючи на всі зусилля, прикладені в періоді новонародженості для вибору відповідної статі виховання, пізніше може виникнути необхідність змінити стать, виходячи з інтересів дитини.
Що таке невизначені (двозначні) геніталії?
Невизначені геніталії (синоніми: двозначні геніталії, інтерсексуалізм, інтерсексуальність, інтерсекс, гермафродитизм (від імен грецьких богів – Гермеса та Афродити)) – це зовнішні статеві органи, що мають ознаки як жіночих, так і чоловічих. Так, наприклад, може бути важко відрізнити збільшений клітор від пеніса, тобто чоловічого статевого члена. Порушення статевої диференціації слід запідозрити не тільки у випадку виражено двозначних статевих органів, але також у випадку переважно чоловічого їх вигляду, але за відсутності у доношеного новонародженого яєчок в калитці з обох сторін, з гіпоспадією (тобто такою вадою розвитку, коли сечовивідний канал не доходить до верхівки пеніса, а його отвір знаходиться будь-де на статевому члені) в поєднанні з відсутністю яєчка в калитці з однієї сторони чи дуже малими розмірами статевого члена.
Порушення статевої диференціації слід запідозрити також у випадку переважно жіночого вигляду геніталій з частковим зрощенням статевих губ, збільшенням клітора або утвореннями, що промацуються в товщі великих статевих губ чи паховій ділянці.
Чому трапляється порушення статевого розвитку та народжуються діти з невизначеними геніталіями?
Всі випадки аномального статевого розвитку поділяються на дві великі групи:
- Помилки первинного визначення статі.
- Помилки статевої диференціації.
До першої групи належать аномалії з боку статевих хромосом та порушення розвитку статевих залоз.
До другої групи належать:
- недостатня маскулінізація (тобто, набуття чоловічих рис) генетичного чоловіка;
- вірилізація (набуття чоловічих рис) генетичної жінки.
Помилки статевої диференціації спричинені переважно гормональними розладами, найчастіше спадкової природи, що виникають у плода під час вагітності та можуть продовжуватися після народження дитини. Крім порушення продукції гормонів може мати місце нечутливість тканин до цих гормонів.
До 6 тижнів вагітності як у чоловічих, так і у жіночих ембріонів розвивається недиференційована тканина статевих залоз і структури статевих органів мають потенціал для розвитку в напрямі обох статей. Зовнішній вигляд геніталій новонародженого у великій мірі визначається присутністю генетичних (Y-хромосома) та гормональних (тестостерон) впливів, що скеровують статеву диференціацію по чоловічому шляху. При відсутності цих впливів статеві органи плода мають схильність розвиватися за жіночим типом. Інтерсексуальний розвиток спричиняється або порушеннями на чоловічому шляху розвитку, які призводять до неповної маскулінізації плоду, або ж вірилізуючим впливом гормонів на розвиток ембріона жіночої, за набором хромосом, статі.
Як визначити стать новонародженої дитини з невизначеними геніталіями?
Визначення статі виховання залежить, в більшій мірі, від анатомії статевих органів та функції статевих залоз і статевих гормонів, а також, в меншій мірі, від набору статевих хромосом. Необхідно не тільки враховувати рівень статевих гормонів у крові, але й здатність тканин реагувати на них. Необхідно повідомити лікарів про захворювання матері та можливий прийом нею ліків, зокрема, гормональних препаратів, під час вагітності.
При визначенні статі дитини враховують наступні фактори:
- здатність до репродуктивної функції (тобто здатність до запліднення та вагітності);
- здатність до нормальної статевої функції; розмір фалюса (статевого члена) має велике значення для можливості розвитку його в повноцінно функціонуючий пеніс під час періоду статевого дозрівання; навпаки, вираженість гіпоспадії не має вирішального значення для визначення статі виховання;
- здатність статевих залоз продукувати відповідні гормони;
- порушення розвитку статевих залоз несе в собі небезпеку злоякісного їх переродження; тому недорозвинені статеві залози у вигляді тяжів здебільшого необхідно усувати хірургічно;
- набір статевих хромосом (генетичні дослідження, визначення каріотипу);
- будову внутрішніх статевих органів;
- дію текстостерону на мозок; дуже високий рівень чоловічого статевого гормону тестостерону в крові плода генетичної дівчинки призводить не тільки до вірилізації зовнішніх статевих органів, але й формує зв’язки між нервовими клітинами мозку таким чином, що ці особи можуть виявляти чоловічі особливості поведінки.
В окремих випадках вдало визначити стать виховання дитини може бути досить складно, і для цього необхідно залучати команду різних спеціалістів та використовувати багато додаткових методів обстеження, щоб вирішити це завдання якнайкраще в стислі терміни. Лікуючий лікар, в разі потреби, може пояснити членам родини суть проблеми. Бажана участь батьків в обговоренні статі виховання дитини. Для того, щоб оцінити дію статевих гормонів, наприклад, здатність тестостерона збільшити розміри статевого члена, потрібне спостереження протягом кількох місяців. У випадку відсутності ефекту може виникнути необхідність перевизначення статі.
Які можливості лікування дітей з невизначеними геніталіями? Планування хірургічної корекції
Хірургічна корекція статевих органів спрямована на усунення чи зменшення їх невизначеності та забезпечення задовільної статевої функції. Якщо діти виховуються як дівчатка, їм звичайно проводять зменшення клітора хірургічним шляхом, здебільшого на 1-му році життя. Збільшення піхви проводять дещо пізніше. Якщо у дитини встановлено нечутливість до чоловічих статевих гормонів або порушення розвитку яєчок і вони мають дуже маленький статевий член, яєчка необхідно усувати і визначати стать виховання як жіночу. Корекцію гіпоспадії проводять звичайно в ранньому віці.
Хірургічна корекція виключно з косметичних міркувань, тобто з метою надати зовнішнім статевим органам “нормального” вигляду, не є невідкладною, незаперечною чи обов’язковою. Її можна відкласти до препубертатного чи пубертатного періоду (періоду статевого дозрівання) та залучити до прийняття рішення саму дитину.
Діти, що народжуються з невизначеними геніталіями, можуть вимагати тривалої гормональної терапії. Так, пацієнти з вродженою гіперплазією наднирників отримують гормональну терапію пожиттєво з моменту встановлення діагнозу. Тривала замінна терапія статевими гормонами розпочинається звичайно в періоді статевого дозрівання.
Особливості виховання дітей, що народилися з невизначеними геніталіями. Психологічні проблеми
Раннє визначення статі та хірургічна корекція геніталій, а також послідовне виховання відповідно до визначеної статі дозволяє мінімізувати психологічні проблеми. Діти, народжені з невизначеними геніталіями, повинні бути під тривалим спостереженням не тільки педіатра, але й дитячого ендокринолога, дитячого уролога, дитячого гінеколога. При виникненні психологічних проблем необхідно вдаватися за допомогою до психолога та психіатра.
В окремих випадках, коли визначення статі може викликати обгрунтовані сумніви, може бути доцільним відтермінувати хірургічну корекцію до початку періоду статевого дозрівання, щоб врахувати відчуття самої дитини при виборі статі.
Психосоціальну кризу в родині, пов’язану з народженням немовляти з невизначеними геніталіями, допомагає спілкування з батьками інших таких дітей. Дана ситуація не є причиною того, щоб соромитися. Таких дітей слід розглядати як особливих, а не як ненормальних. Велику допомогу можуть надати громадські товариства підтримки.
Переглянуто редакційною колегією I.B.I.S.: 27/08/2003
Дивіться також:
- Генетична регуляція статі та діагностичні підходи при інтерсексуалізмі (інформація для спеціалістів)
- Новонароджена дитина з невизначеними (двозначними) геніталіями (інформація для спеціалістів)
Новонароджена дитина з невизначеними (двозначними) геніталіями
(Intersex – Hermaphroditism)
Ю.С. Коржинський
Доктор медичних наук, професор
Завідувач кафедри педіатрії і неонатології
Львівського державного медичного університету
Народження дитини з невизначеними геніталіями є однією із складних медичних та соціальних проблем, що ставить ряд складних завдань перед лікарями та болюче сприймається батьками. Статева невизначеність новонародженого немовляти нерідко перетворюється в соціальну кризу. Це вимагає швидкого і в той же час докладного та поглибленого обстеження немовляти перед тим, як визначиться стать дитини та розпочнеться лікування. Обстеження такої дитини і визначення її статі вимагає участі лікарів різних спеціальностей та використання ряду додаткових методів діагностики. Воно повинно бути проведено так швидко, як це можливо, але, в той же час, зайняти стільки часу, скільки потрібно. У більшості випадків після народження такої дитини виникає потреба переведення її в спеціалізоване відділення, де є найкращі умови для проведення обстеження.
В цілому ряді випадків визначення статі новонародженої дитини з невизначеними геніталіями може бути дуже складним завданням, і до обговорення при прийнятті остаточного рішення варто залучати батьків дитини. Кожен такий випадок вимагає індивідуального підходу. Рекомендується не реєструвати народження дитини і не давати дитині ім’я, якщо можливо, доки не визначено стать виховання. Необхідно усвідомлювати, що в окремих випадках, незважаючи на всі зусилля, прикладені в періоді новонародженості для вибору відповідної статі виховання, пізніше може виникнути необхідність змінити стать, виходячи з інтересів дитини.
Визначення:
Невизначені геніталії (синоніми: двозначні геніталії, інтерсексуалізм, інтерсексуальність, інтерсекс, гермафродитизм (від імен грецьких богів – Гермеса та Афродити)) – це зовнішні статеві органи, що мають ознаки як жіночих, так і чоловічих. Так, наприклад, може бути важко відрізнити збільшений клітор від пеніса.
Порушення статевої диференціації слід запідозрити не тільки у випадку виражено двозначних статевих органів, але також у випадку переважно чоловічого їх вигляду, але за відсутності у доношеного новонародженого яєчок в калитці з обох сторін, гіпоспадією в поєднанні з відсутністю яєчка в калитці з однієї сторони чи дуже малими розмірами статевого члена.
Порушення статевої диференціації слід запідозрити також у випадку переважно жіночого вигляду геніталій з частковим зрощенням статевих губ, збільшенням клітора або утвореннями, що промацуються в товщі великих статевих губ чи паховій ділянці (табл 1).
Клінічні симптоми у новонародженого, що вказують на можливість
порушення статевої диференціації
геніталії |
|
геніталії |
|
геніталії |
|
Класифікація, етіологія та патогенез:
Класифікація порушень статевого розвитку
(J.S.D. Winter, R.M. Couch, 1995)
- Помилки первинної детермінації статі
- Аномалії статевих хромосом
- З яєчками і чоловічим фенотипом
- Синдром Клайнфельтера (47, XXY та інші варіанти)
- Синдром XYY
- ХХ чоловіки (Х-Y взаємообмін)
- З яйниками і жіночим фенотипом
- Трісомія Х і варіанти
- З бісексуальними ґонадами
- Справжній гермафродитизм (Х-Y взаємообмін)
- Химеричний (ХХ/XY) справжній гермафродитизм
- Мозаїчний (ХХ/XХY) справжній гермафродитизм
- З дизґенетичними ґонадами
- Аномалії Y-хромосоми (ХYр)
- Змішана дизґенезія гонад (45, Х/46, ХY)
- Синдром Тернера (45, Х та варіанти)
- З яєчками і чоловічим фенотипом
- Аномалії ґонадоґенезу
- Порушення розвитку яйників
- ХХ дизґенезія гонад
- Порушення розвитку яєчок
- ХY дизґенезія гонад
- Дизґенетичний чоловічий псевдогермафродитизм
- Синдром регресії яєчок (аґонадизм, рудиментні яєчка, анорхія)
- Порушення розвитку яйників
- Аномалії статевих хромосом
- Порушення статевої диференціації
- Неадекватна маскулінізація генетичного чоловіка
- Недостатня регресія парамезонефричної протоки
- Персистуюча парамезонефрична протока
- Недостатня ґенітальна вірилізація
- Гіпоплазія клітин Лейдіґа (дефект рецептора LH/hCG)
- Вроджені порушення синтезу кортикостероїдів і тестостерона
- Дефіцит 20, 22-десмолази
- Дефіцит 3b-гідроксистероїд дегідроґенази
- Дефіцит 17a-гідроксилази
- Вроджені порушення синтезу тестостерона
- Дефіцит 17, 20-десмолази
- Дефіцит 17b-гідроксистероїд дегідрогенази
- Вроджене порушення метаболізму тестостерона
- Дефіцит 5a-редуктази
- Дефект відповіді клітин-мішеней
- Повна нечутливість до андроґенів
- Неповна нечутливість до андроґенів
- Фемінізація, спричинена зовнішнім середовищем
- Прийом матір’ю естроґенів або прогестина
- Мультиґенна/мультифакторіальна
- Ізольовані гіпоспадії і крипторхізм
- Множинні вроджені аномалії
- Недостатня регресія парамезонефричної протоки
- Вірилізація ґенетичної жінки
- Вроджена гіперплазія наднирників
- Дефіцит 21-гідроксилази
- Дефіцит 11b-гідроксилази
- Дефіцит 3b-гідроксистероїд дегідроґенази
- Гормони зовнішнього середовища
- Вірилізуючі захворювання матері
- Ятроґенна вірилізація
- Тератологічні вади розвитку
- Аномальний розвиток піхви або матки
- Синдром множинних вроджених аномалій
- Вроджена гіперплазія наднирників
- Неадекватна маскулінізація генетичного чоловіка
До 6 тижнів вагітності як у чоловічих, так і у жіночих ембріонів розвивається недиференційована тканина статевих залоз і структури статевих органів мають потенціал для розвитку в напрямі обох статей. Зовнішній вигляд геніталій новонародженого у великій мірі визначається присутністю генетичних (Y-хромосома) та гормональних (тестостерон) впливів, що скеровують статеву диференціацію по чоловічому шляху (табл.3). При відсутності цих впливів статеві органи плода мають схильність розвиватися за жіночим типом (рис.1).
Рис. 1. Роль гонадальних гормонів у розвитку статевих фенотипів
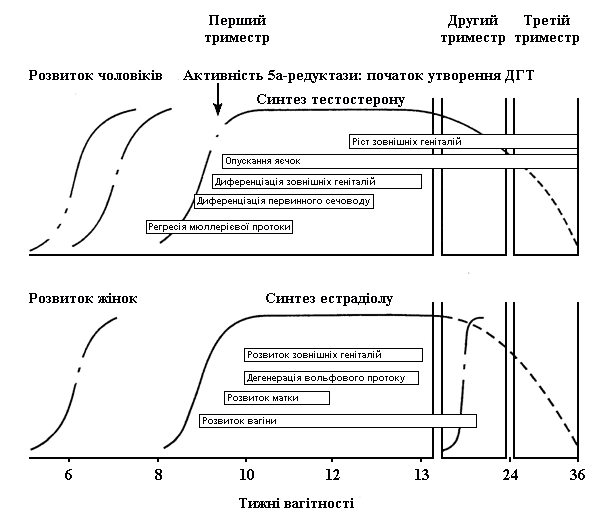
Інтерсексуальний розвиток спричиняється або порушеннями на чоловічому шляху розвитку, що призводять до неповної маскулінізації плоду, або ж вірилізуючим впливом гормонів на розвиток ембріона жіночої, за набором.
Послідовність статевого розвитку
| Первинні зародкові клитини починають мігрувати до генітального гребня | |
| Генітальний гребінь формує недиференційовану гонаду | |
| Поява мюллерових проток; розвинення яєчок | |
| Активізується мюллерів інгібітор (з яєчок) | |
| Починається синтез тестостерону (викликаний плацентарним хоріонним гонадотропіном) | |
| Злиття лабіоскротальної припухлості | |
| Закриття медіальної борозди | |
| Закриття уретральної борозди | |
| Завершення регресії мюллерієвої протоки |
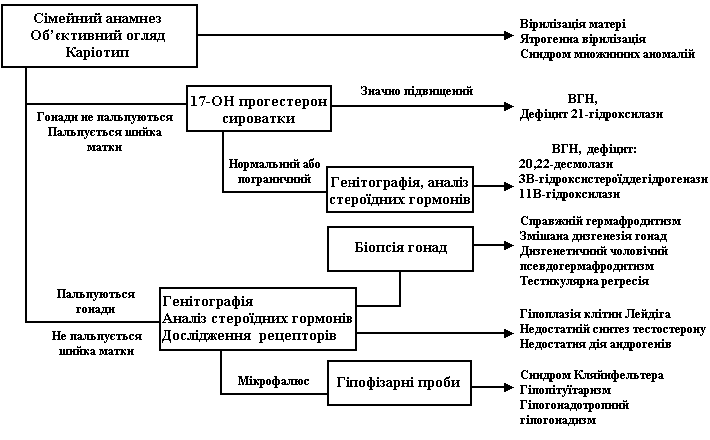
Діагностика та диференційна діагностика:
Об’єктивний огляд новонароджених з невизначеними геніталіями. Зовнішній вигляд геніталій не є специфічним для окремих нозологічних одиниць. При огляді необхідно зосередити увагу на наступних ознаках. Необхідно виміряти довжину фалюса (рис. 2), відзначити положення отвору уретри, кількість і розташування вентральних вуздечок на фалюсі, ступінь можливого зрощення статевих губ чи розщеплення калитки, наявність або відсутність гонад, які можна пропальпувати, їх консистенцію, розмір та локалізацію.
Рис. 2. Діаграми антропометрії статевих органів новонароджених
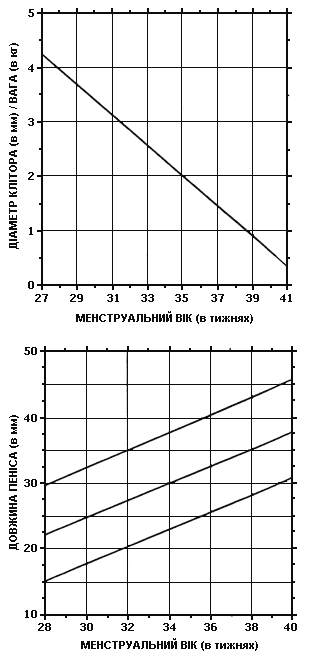
Будь-яка гонада, що пропальповується в лабіоскротальній або паховій (пахвинній) ділянці, містить тканину яєчка. Якщо окремі ділянки гонади відрізняються за консистенцією, можна запідозрити овотестіс або неопластичну трансформацію гонади.
Обережне бімануальне ректальне дослідження дозволить виявити шийку матки по середній лінії. Слід звернути увагу на можливі поєднані аномалії. Зокрема, синдром, що включає пухлину Вільмса, гемігіпертрофію, аніридію, двозначні геніталії та гонадобластому може виявляти автосомально-домінантний тип спадкування або проявлятися спорадично, внаслідок делеції 11р13.
Необхідно звернути увагу на те, чи присутні ознаки дегідратації або ділянки гіперпігментації. Останні пов’язані з хронічною недостатністю кори наднирників, гіперпродукцією АКТГ та можуть вказувати на вроджену гіперплазію наднирників (ВГН).
Збір анамнезу. Необхідно вияснити, чи не було в родині випадків гіпоспадії, крипторхізму, неплідності (бездітні тітки), а також, чи не було кровних шлюбів, що підвишують імовірність автосомно-рецесивних захворювань. Необхідно розпитати, чи мати не приймала медикаментів під час вагітності, зокрема, таких, як прогестерон, даназол, тестостерон, дифенін, аміноглютетимід. Розпитати, чи не спостерігалося вірилізації матері під час вагітності. Смерть попередніх дітей в неонатальному періоді може вказувати на вроджену гіперплазію наднирників. Плацентарна недостатність через хоріонічний гонадотропін людини впливає на синтез тестостерону у яєчках плода.
Додаткові методи обстеження, що застосовуються для диференціювання різних причин невизначених геніталій:
- Хромосомний аналіз (каріотипування). Якщо результати каріотипування відомі з затримкою в кілька днів, ці дослідження доцільно доповнити визначенням статевого хроматину, що дає швидкий результат.
- Дослідження рівня стероїдних гормонів в крові та екскреції їх з сечею, в т.ч. дослідження здатності яєчка продукувати тестостерон у відповідь на стимуляцію хоріонічним гонадотропіном.
- Дослідження здатності тканин реагувати на тестостерон.
- Ультразвукове дослідження органів малого тазу.
- Комп’ютерна томографія наднирників.
- Генітографія (введення контрасту в отвір сечовипускного каналу, уретроцистографія). Цей метод може візуалізувати вагіну, матку та фалопієві труби, якщо вагіна виходить з урогенітального синуса.
- Застосування магнітно-ядерного резонанса дозволяє локалізувати яєчка в черевній порожнині.
Послідовність проведення параклінічних досліджень та диференціально-діагностичних процедур залежить від того:
- чи можна пропальпувати гонади;
- який каріотип немовляти.
Отже, диференціальний діагноз починають з отримання відповіді на ці питання. Якщо гонади не пальпуються, визначають рівень 17-гідроксипрогестерону в сироватці крові. Його високий рівень у ХХ-немовляти говорить на користь вродженої гіперплазії наднирників, спричиненої дефіцитом 21-гідроксилази, що є найчастішою причиною вірилізації ХХ плода та невизначених геніталій у новонароджених взагалі. Необхідно слідкувати за масою новонародженої дитини, об’ємом засвоєної нею рідини. Електроліти сироватки крові треба визначати щоденно з метою раннього виявлення синдрому втрати солі. Визначення рівня реніну плазми слід проводити не раніше 4-го дня. Крім підвищення 17-гідроксипрогестерону сироватки та реніну плазми, при дефіциті 21-гідроксилази спостерігається також підвищення рівня тестостерону сироватки. Якщо рівень 17-гідроксипрогестерону не перевищує 300 нг/л, необхідно визначити інші стероїди в сироватці, такі, як 11-деоксикортизол, дегідроепіандростерон, та 17-гідроксипрегненолон, що допоможе виявити інші форми ВГН та жіночого псевдогермафродитизму.
Якщо результати стероїдів нормальні, ультразвукове дослідження яйників або комп’ютерна томографія наднирників може виявити маскулінізуючу пухлину. Лапаратомія та біопсія гонади можуть бути необхідними для діагностики справжнього гермафродитизму.
Якщо у немовляти пальпуються гонади, вони містять тестикулярну тканину. Але навіть якщо каріотип є ХY, не треба спішити називати дитину хлопчиком.
Якщо у немовляти пальпуються гонади, вони містять тестикулярну тканину. Але навіть якщо каріотип є ХY, не треба спішити називати дитину хлопчиком.
Існують різні варіанти дизгенезій гонад. Так, при каріотипі ХY може розвинутися “чиста” дизгенезія гонад, причиною якої є аномалії гена SRY (фактор, що визначає яєчко). Немовлята не можуть маскулінізуватися через неповну диференціацію яєчка. Геніталії переважно жіночі, але можлива кліторомегалія. Пальпуються гонади у вигляді тяжів. У 30% випадків розвивається гонадобластома або дизгермінома, тому гонади необхідно усувати в немовлячому періоді. Стать визначається як жіноча.
При мозаїчному каріотипі ХО/ХY (45,Х/46 ХY) розвивається змішана дизгенезія гонад. З одного боку, звичайно, присутній яйник, з іншого – яєчко. Гонади дизгенетичні. З огляду на високий ризик їх злоякісного переродження, гонади у вигляді тяжів необхідно усувати у віці немовляти. Зовнішні статеві органи можуть бути від переважно чоловічих до цілком жіночих, звичайно присутні матка та фалопієві труби. Стимуляція хоріонічним гонадотропіном людини на протязі 3-х днів дозволяє оцінити здатність яєчка продукувати тестостерон. Дані цього тесту є одним з аргументів щодо вибору статі та усунення яєчка.
При справжньому гермафродитизмі 10% пацієнтів мають каріотип ХY, 10% – мозаїчний, а 80% – ХХ. Визначення статі залежить від анатомії зовнішніх та внутрішніх статевих органів.
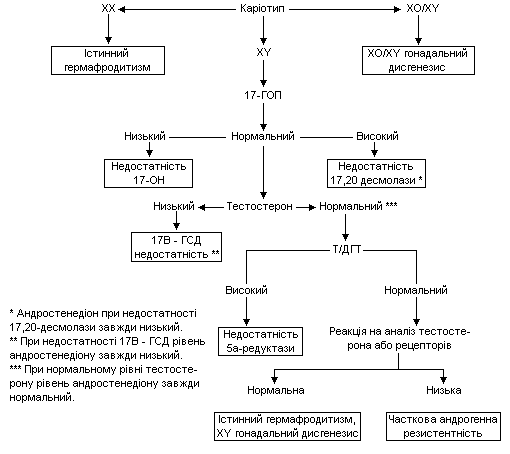
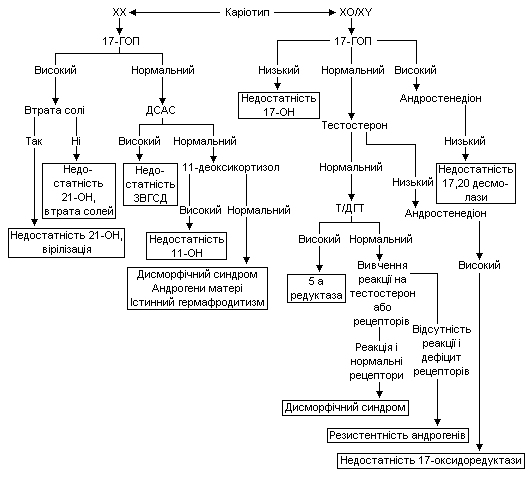
Причини та підстави лабораторної діагностики недостатньої маскулінізації ХY немовляти подано в таблиці 4.
Таблиця 4.
Недостатня маскулінізація генетично чоловічої статі

Запропоновано ряд діагностичних алгоритмів при народженні немовлят з невизначеними геніталіями, від значно спрощених до більш деталізованих.
Рис. 3. Спрощений алгоритм обстеження та діагностики новонародженої дитини з невизначеними геніталіями
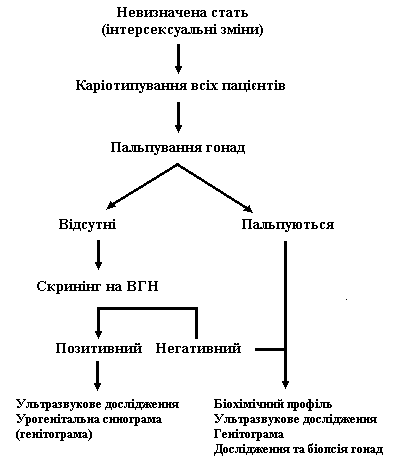
Рис. 4. Спрощений алгоритм обстеження та діагностики новонародженої дитини з невизначеними геніталіями
Гонади пальпуються
Гонади не пальпуються
ГСД – гадроксистероїддегідрогеназа
Т/ДГТ – співвідношення тестостерона до дегідротестостерона
ГОП – гідроксипрогестерон
11-ОН – 11-гідроксилаза
17-ОН – 17-гідроксилаза
21-ОН – 21-гідроксилаза
Визначення cтаті немовляти з невизначеними геніталіями:
Визначення cтаті немовляти з невизначеними геніталіями є не менш складним і відповідальним завданням, ніж діагностика причини та нозологічної форми порушення статевої диференціації. Щодо деяких станів, на сьогоднішній день існують суперечливі рекомендації. Виділяють наступні різновиди статі:
- генетична стать (визначається статевими хромосомами);
- генетична стать (визначається статевими залозами);
- генітальна стать (визначається будовою та зовнішнім виглядом зовнішніх статевих органів);
- гормональна стать (визначається рівнем статевих гормонів в крові);
- гендерна стать (визначається вихованням, але також самоідентифікацією. Остання може залежати від внутрішньутробної дії тестостерона на ЦНС);
- документальна стать зазначається в офіційних документах.
Цілком очевидно, що без порушень статевої диференціації всі ці різновиди статі співпадають між собою. У випадку порушення статевої диференціації, навпаки, вони як правило, не співпадають.
Визначення статі виховання залежить, в більшій мірі, від анатомії статевих органів та функції статевих залоз і статевих гормонів, а також, в меншій мірі, від набору статевих хромосом. Необхідно не тільки враховувати рівень статевих гормонів у крові, але й здатність тканин реагувати на них. Для того, щоб оцінити дію статевих гормонів, наприклад, здатність тестостерона збільшити розміри статевого члена, потрібне спостереження протягом кількох місяців. У випадку відсутності ефекту може виникнути необхідність перевизначення статі.
При визначенні статі дитини враховують наступні фактори:
- здатність до репродуктивної функції;
- здатність до нормальної статевої функції. Розмір фалюса має велике значення для можливості розвитку його в повноцінно функціонуючий пеніс під час періоду статевого дозрівання. Навпаки, вираженість гіпоспадії не має вирішального значення для визначення статі виховання;
- здатність статевих залоз продукувати відповідні гормони;
- порушення розвитку статевих залоз несе в собі небезпеку злоякісного їх переродження. Тому недорозвинені статеві залози у вигляді тяжів здебільшого необхідно усувати хірургічно;
- набір статевих хромосом (генетичні дослідження, визначення каріотипу;
- будову внутрішніх статевих органів;
- дію текстостерону на мозок. Дуже високий рівень чоловічого статевого гормону тестостерону в крові плода генетичної дівчинки призводить не тільки до вірилізації зовнішніх статевих органів, але й формує міжнейрональні зв’язки в ЦНС таким чином, що ці особи можуть виявляти чоловічі особливості поведінки, інакше кажучи, демонструвати гінекофільну поведінку.
Враховуючи, що всі генетичні дівчатка з вродженою гіперплазією наднирників потенційно фертильні, їх рекомендують виховувати як дівчаток. Однак, M. Diamond рекомендує генетичних дівчаток з високим ступенем вірилізації виховувати як хлопчиків, враховуючи дію тестостерона на ЦНС плода, що може спричинити гінекофільну поведінку в майбутньому. Чим вищий ступінь вірилізації, тим більший вплив тестостерона на формування міжнейронних зв’язків.
Якщо існують і матка, і вагіна, а зовнішні геніталії потребують декількох стадій відновлення гіпоспадії, дитину рекомендують виховувати, як дівчинку.
Всі пацієнти з повною нечутливістю до тестостерону (так звана тестикулярна фемінізація) повинні виховуватися як дівчатка, гонади необхідно усувати.
Необхідно бути обережним, рекомендуючи стать виховання, відмінну від генетичної.
В багатьох випадках вдало визначити стать виховання дитини може бути досить складно, для цього необхідно залучати команду спеціалістів, таких, як дитячий ендокринолог, дитячий уролог, спеціаліст з променевої діагностики, дитячий гінеколог, генетик, морфолог. Цю команду очолює звичайно неонатолог, педіатр або дитячий ендокринолог. Спеціаліст, що очолює команду, повинен координувати мультидисциплінарні зусилля та контактувати з батьками дитини, в зрозумілій формі пояснювати їм суть проблеми. Згідно з сучасними поглядами, доцільно залучати батьків до обговорення визначення статі виховання дитини.
Лікування дітей з невизначеними геніталіями:
Хірургічна корекція статевих органів спрямована на усунення чи зменшення їх невизначеності та забезпечення задовільної статевої функції. Якщо діти виховуються як дівчатка, їм звичайно проводять редукцію клітора хірургічним шляхом, здебільшого на 1-му році життя. Сучасна оперативна техніка дозволяє не тільки сконструювати нормальну на вигляд вульву, але й зберегти інервацію клітора і його функціональність. Збільшення піхви проводять дещо пізніше, після 1 року життя. Якщо у дитини встановлено нечутливість до чоловічих статевих гормонів або порушення розвитку яєчок і вони мають дуже маленький статевий член, яєчка необхідно усувати і визначати стать виховання як жіночу. Корекцію гіпоспадії проводять звичайно в ранньому віці.
Хірургічна корекція виключно з косметичних міркувань, тобто з метою надати зовнішнім статевим органам “нормального” вигляду не є невідкладною, незаперечною чи обов’язковою. Її можна відкласти до препубертатного чи пубертатного періоду (періоду статевого дозрівання) та залучити до прийняття рішення саму дитину. Існує навіть досить категорична точка зору щодо мораторію на косметичну хірургічну корекцію геніталій в ранньому віці, з тим, щоби в препубертатному або пубертатному віці врахувати відчуття і бажання самого пацієнта.
Діти, що народжуються з невизначеними геніталіями, можуть вимагати тривалої гормональної терапії. Так, пацієнти з вродженою гіперплазією наднирників отримують гормональну терапію пожиттєво з моменту встановлення діагнозу. Тривала замінна терапія статевими гормонами, наприклад, у випадку усунення гонад, розпочинається звичайно в періоді статевого дозрівання.
Особливості виховання дітей, що народилися з невизначеними геніталіями:
Раннє визначення статі та хірургічна корекція геніталій, а також послідовне виховання відповідно до визначеної статі дозволяє мінімізувати психологічні проблеми. Діти, народжені з невизначеними геніталіями, повинні бути під тривалим спостереженням не тільки педіатра, але й дитячого ендокринолога, дитячого уролога, дитячого гінеколога. При виникненні психологічних проблем необхідно вдаватися за допомогою до психолога та психіатра.
В окремих випадках, коли визначення статі може викликати обгрунтовані сумніви, може бути доцільним відтермінувати хірургічну корекцію до початку періоду статевого дозрівання, щоб врахувати відчуття самої дитини при виборі статі.
Психосоціальну кризу в родині, пов’язану з народженням немовляти з невизначеними геніталіями, допомагає перемогти спілкування з батьками інших таких дітей. Дана ситуація не є причиною того, щоб соромитися. Таких дітей слід розглядати як особливих, а не як ненормальних. Велику допомогу можуть надати громадські товариства підтримки.
Пренатальна діагностика та профілактика:
Якщо в родині були випадки народження інтерсексуальних дітей, необхідно провести медико-генетичне консультування та спробу пренатальної діагностики невизначених геніталій. Ультразвукове дослідження дозволяє візуалізувати геніталії після 20 тижнів вагітності, амніоцентез може забезпечити біохімічну діагностику при 15-20 тижнях гестації, а біопсія ворсинок хоріона дозволяє визначити генетичну стать та провести НLA-типування або ДНК-діагностику ще раніше.
Для захворювання, що є найчастішою причиною невизначених геніталій, а саме, вродженої гіперплазії наднирників (ВГН), спричиненої дефіцітом 21-гідроксилази, розроблено протокол дородової діагностики та профілактики вірилізації генетичної дівчинки шляхом призначення вагітній жінці дексаметазона. Класична форма захворювання зустрічається з частотою 1:10000, стерті некласичні форми зустрічаються з частотою 1:1000. Частота гетерозиготного носійства – 1:60. Якщо в сім’ї вже народилася дитина з ВГН, жінці призначають дексаметазон по 0,5 х 3 рази на день з метою запобігання вірилізації плоду жіночої статі з 4 – 5 тижня вагітності, тобто, як тільки відомо, що вона вагітна. Жінка продовжує приймати препарат до проведення дородової діагностики або за допомогою визначення 17-гідроксипрогестерону та D4-андростенедіону в навколоплідних водах, або за допомогою проведення HLA-типування чи ДНК- діагностики щляхом біопсії ворсин хоріона. Лікування продовжується до народження дитини, якщо плід уражений (діагностовано ВГН) і жіночої статі, що становить 1/8 всіх вагітностей.
Література:
- Evaluation of the newborn with developmental anomalies of the external genitalia (RE99580). American Academy of Pediatrics. Pediatrics 2000;106:138-142.
- Winter JSD, Couch RM. Sexual Differentiation / Endocrinology and Metabolism P. Felig, J. D. Baxter, L. A. Frohman (Ed.).— McGraw-Hill Inc., New York.- 1995:1053-1104.
- Скотт М.Д. Невизначені геніталії /Посібник з неонатології.- Ред.: Д. Клоерті, Е. Старк.- Фонд Допомоги Дітям Чорнобиля. Київ, 2002.- С.581-590.
- Donahoe PK, Schnitzer JJ. Evaluation of the infant who has ambiguous genitalia, and principles of operative management. Semin. Pediatr. Surg. 1996;5:30-40.
- Diamond M, Sigmundson H.K. Sex reassignment at birth: a long term review and clinical implications. Archives of Pediatrics and Adolescent Medicine. 1997;151:298-304.
- Diamond M, Sigmundson H.K. Management of intersexuality: guidelines for dealing with persons with ambiguous genitalia. Archives of Pediatrics and Adolescent Medicine. 1997;151:1046-1050.
- Reiner WG. Sex assignment in the neonate with intersex or inadequate genitalia. Archives of Pediatrics and Adolescent Medicine. 1997;151:1044-1045.
- Diamond M. Pediatric management of ambiguous and traumatized genitalia. The Journal of Urology. 1999;162:1021-1028.
- Forest MNG, Betuel H, Couillin P et al. Prenatal diagnosis of congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency by steroid analysis in the amniotic fluid of mid-pregnancy: Comparison with HLA typing in 17 pregnancies at risk for CAH. Prenatal Diagnosis. 1981;1:197.
- Hughes IA, Laurence KM. Prenatal diagnosis of congenital adrenal hyperplasia due to 21-hydroxylase deficiency by amniotic fluid steroid analysis. Prenatal Diagnosis. 1982;2:97.
- Pang S, Pololack MS, Marshall RN, Immken L. Prenatal treatment of congenital adrenal hyperplasia due to 21-hydroxylase deficiency. New England Journal of Medicine. 1990;322:111.
- Laue L, Cutler GB, Jr. 21-hydroxylase deficiency: Overview of treatment. The Endocrinologist. 1992;2:291.
- Саул Р., Гір Д., Сівер Л., Фелан М., Світ К., Міллс К. Довідник росту від третього триместру до підліткового віку /Грінвудський генетичний центр. Університет Південної Алабами. ”Вісник” Київ-Рівне-Луцьк-Хмельницький-Херсон-Симферополь, 2002.
- Sultan C, Paris F, Lumbroso S, Galifer RB. Ambiguous genitalia in the newborn. Seminars in Reproductive Medicine. 2002;20:181-188.
Переглянуто редакційною колегією I.B.I.S.: 02/09/2003
Дивіться також:
- Генетична регуляція статі та діагностичні підходи при інтерсексуалізмі (інформація для спеціалістів)
- Новонароджена дитина з невизначеними (двозначними) геніталіями (інформація для батьків)
|
|
|
|
|
|






{kind=link}