
Uncategorized
Освіта дітей дошкільного та раннього шкільного віку з ФАС та частковим ФАС

Посібник для вчителя підготовлений спеціалістами Центру дослідження фетального алкогольного синдрому (ФАС) Інституту Маркуса, Атланта, Штат Джорджія, США (Marcus Institute, Atlanta, GA 30329). Профінансовано Центром з контролю та запобігання захворюванням, гранти U84/CCU320162-02 та U01-DD000039-02.
Переклад з англійської та адаптацію українською мовою здійснено за підтримки МБФ “ОМНІ-мережа для дітей”. Редагування україномовної версії: практичний психолог РОКЛДЦ ім. В. Поліщука Грановська І.В.
ЗМІСТ:
- Розділ 1: Що таке ФАС/частковий ФАС?
- Розділ 2: Як алкоголь впливає на пізнавальну здатність та розвиток?
- Розділ 3: Порушення нервового розвитку, які впливають на вивчення математики.
- Розділ 4: Інтерактивне вивчення математики за допомогою програми MILE.
- Розділ 5: Як допомогти в навчанні дітям з ФАС.
- Розділ 6: Засоби та стратегії подолання порушень нервового розвитку, які впливають на вивчення математики.
- Розділ 7: Середовище в класі, оптимальне для дітей з ФАС.
- Розділ 8: Поширені запитання.
- Додаток: Приклад інтерактивного навчального діалогу, заснованого на матеріалі одного з уроків програми MILE.
Пренатальний – неонатальний розвиток
Вік та життєздатність. 26-тижневий гестаційний вік (кількість тижнів з початку останнього менструального циклу до народження) визначається як початок періоду життєздатності. Читач зрозуміє, що це положення є відносним, так як ні вага, ні довжина, ні вік плоду не можуть бути абсолютними для визначення життєздатності. За останні три десятиліття менструальний вік, при якому дитина може жити поза маткою, значно зменшився завдяки досягненням у інтенсивній терапії новонароджених. У цьому розділі подаються дані про ріст та розвиток плоду з початку періоду життєздатності до неонатального періоду.
Вимірювання. Не існує фіксованого набору вимірів, які можна було б застосувати до усіх людей. Наскільки це можливо, ми будемо давати відомості про населення, вимірююче яке, ми створили дані діаграми. В деяких випадках, при наявності більше, ніж одного джерела, надаватимуться різні дані, що дозволить їх порівняння. У розділі “Додаткові рекомендації” подаються додаткові специфічні расові та популяційні дані.
Параметри росту новонароджених. Розмір новонароджених залежить від багатьох чинників, одним із найголовніших серед них є тривалість вагітності. В цьому розділі вона визначається як менструальний вік (гестаційний вік), тобто кількість тижнів від початку останнього менструального циклу до народження. Менструальний вік на два тижні довший за овуляційний. Усі вимірювання також співставляються з вагою при народженні та постнатальним віком.
Спочатку наводяться загальні параметри росту та розвитку дитини, далі черепно-лицьові виміри та виміри тулуба і кінцівок. Завершують розділ відомості про постнатальний розвиток недоношених дітей.
- Ріст та розвиток
- Ріст плоду
- Осифікація
- Прикріплення та довжина пуповини
- Вага при народженні та гестаційний вік (на рівні моря)
- Вага при народженні та гестаційний вік (1610 метрів над рівнем моря і вище)
- Зріст при народженні та гестаційний вік (на рівні моря)
- Зріст при народженні та гестаційний вік (1610 метрів над рівнем моря і вище)
- Обвід голови при народженні та гестаційний вік (на рівні моря)
- Обвід голови при народженні та гестаційний вік (1610 метрів над рівнем моря і вище)
- Тім’ячково-куприкова довжина
- Передньо-задній діаметр голови
- Біпарієтальний діаметр голови
- Вимірювання відстані між очима
- Довжина очних щілин
- Довжина очних щілин (продовження)
- Вимірювання відстані між зіницями
- Вимірювання довжини вуха
- Розміщення вуха відносно очей
- Розміщення вуха відносно очей (продовження)
- Ширина рота
- Довжина фільтру
- Виміри тулуба
- Відстань між сосками
- Довжина тулуба
- Довжина грудини
- Вимірювання верхніх кінцівок
- Довжина верхньої кінцівки
- Вимірювання плеча
- Вимірювання передпліччя
- Вимірювання кистей та нігтів пальців рук
- Вимірювання середнього пальця
- Вимірювання тулуба і нижніх кінцівок
- Вимірювання гомілки
- Довжина нижніх кінцівок
- Довжина ступні
- Довжина та діаметр пеніса
- Вимірювання геніталій
- Постнатальний розвиток недоношених дітей
Фетальний алкогольний синдром (ФАС) – одна із найпоширеніших причин вроджених вад і порушень розвитку дітей
(Fetal Alcohol Syndrome, FAS)
Ірина Грановська
практичний психолог
обласного медико-генетичного центру РОКЛДЦ ім. В. Поліщука
9 вересня – Міжнародний день запобігання фетальному алкогольному синдрому (ФАС).
Відзначення цієї дати започатковане у 1999 році з метою нагадування суспільству, що вплив алкоголю під час вагітності – одна із основних причин вроджених вад і порушень у розвитку дітей. ФАС спостерігається навіть частіше, ніж синдром Дауна. Чому ж так? За даними літератури, понад 80% жінок вживають алкоголь до вагітності, а від 3 до 20% не відмовляються від нього, виношуючи дитину. Дослідження свідчать, що жінки не завжди володіють достатньо інформацією про шкоду, яку завдає алкоголь під час вагітності на розвиток дитини, а тому і не виключають вживання алкоголю під час вагітності.
Мозок, що розвивається, є найбільш вразливою до пренатального впливу алкоголю структурою.
Не існує єдиної безпечної дози, оскільки шкідливість алкоголю також залежить від особливостей жіночого організму.
Отже, ФАС виникає, коли дитина зазнає впливу алкоголю, знаходячись в утробі матері, тобто ще до народження (пренатально). Слід зауважити, що не всі діти, чиї матері вживали алкоголь під час вагітності, мають ФАС. ФАС виникає у дітей близько 40% матерів, що зловживали алкоголем.
ФАС – це узагальнююча назва для різних за ступенем тяжкості проявів відхилень у розвитку дитини. Більш точним, є використання терміну «фетальний алкогольний спектр порушень – ФАСП». Вважають, що частота ФАСП у дітей складає 1%. ФАСП охоплює весь діапазон можливих наслідків впливу алкоголю у пренатальному періоді. Він включає:
- Фетальний алкогольний синдром (ФАС) – найважчий наслідок пренатального впливу алкоголю. Частота ФАС серед дітей у США становить 0,5-2 випадки, а в Україні – 0,5-0,6 на 1000 новонароджених.
- Частковий фетальний алкогольний синдром (чФАС) – означає, що присутні не всі критерії для діагнозу ФАС або ж наслідки пренатального впливу алкоголю є менш важкими.
- Пов’язане з алкоголем порушення нейророзвитку (ПАПН) – коли відсутні явні фізичні ознаки, які вказують на пренатальний вплив алкоголю, але підозрюється ураження мозку.
ФАС та чФАС – це медичні діагнози, які встановлюються лікарем (неонатологом, педіатром, генетиком), який пройшов спеціальне навчання. Діагноз базується на чотирьох критеріях:
- Пренатальний вплив алкоголю – підтверджено вживання алкоголю під час вагітності.
- Обличчя – специфічні риси обличчя.
- Фізичний розвиток – мала маса тіла при народженні та/чи повільний постнатальний ріст.
- Мозок – ушкодження центральної нервової системи.
Більшість проявів ФАС є вторинними наслідками дії алкоголю на розвиток мозку.


У дітей, що зазнали пренатального впливу алкоголю, часто спостерігаються специфічні риси обличчя. На малюнках для порівняння схематично показано дитину з ознаками ФАС та дитину загального розвитку, у якої цих рис немає. Слід звернути увагу на розмір голови, форму губ та очі.
Ось характерні для ФАС особливості голови та обличчя:
| Голова | Очі | Вуха | Рот |
|---|---|---|---|
|
|
|
|
Діти з ФАС мають меншу масу тіла при народженні. Крім того, вони можуть бути меншого зросту, ніж інші новонароджені. Більшість уражених алкоголем дітей залишаються меншими за однолітків протягом усього дитинства. У підлітковому віці вони інколи можуть «наздогнати» інших у рості. Але все ж більшість уражених алкоголем підлітків та дорослих мають зріст нижче середнього.
Крім того, діти з ФАС мають проблеми нейророзвитку. Під «нейророзвитком» мається на увазі те, як мозок дитини розвивається з часом. У дітей, які зазнали значного впливу алкоголю у пренатальному періоді, можуть бути зміни фізичної структури мозку. Ці зміни можуть мати досить широкий діапазон проявів. Все залежить від локалізації та міри ураження мозку. До найбільш поширених наслідків пренатального впливу алкоголю на нейророзвиток належать:
- Затримка розвитку чи розумова відсталість;
- Порушення уваги та проблеми з контролем збудження – однак, не класичний синдром дефіциту уваги з гіперактивністю;
- Проблеми з навчанням або пізнавальні розлади – особливо у сфері математики;
- Порушення розвитку мови та мовлення – найбільше проявляються у вживанні комплексного мовлення та поганому розумінні змісту абзаців;
- Специфічні проблеми з критикою та соціальною поведінкою;
- Проблеми з моторикою;
- Візуально-просторові порушення.
Тому дітям з ФАСП, щоб осягнути свій потенціал потрібна своєчасна освітня або корекційна підтримка. Інколи це означає, що такі діти відповідають критеріям для отримання спеціальних ранніх освітніх послуг.
Слід зазначити, що з 2003 року у співпраці з Програмою запобігання вродженим вадам розвитку ОМНІ-мережі та CIFASD (Спільною ініціативою з дослідження фетального алкогольного спектру порушень) в Рівненській області проведено низку освітніх заходів для медичних працівників, психологів і широкого загалу з питань діагностики, раннього втручання і запобігання ПФАС. Своєчасне розпізнавання порушень розвитку, спричинених пренатальним впливом алкоголю, сприяє, зокрема, застосуванню спеціальних навчальних програм, які дають шанс дитині досягти кращих результатів у поведінці та розвитку.
Наприклад, програма з інтерактивного вивчення математики «MILE», розроблена фахівцями Центру з дослідження ФАС університету Еморі (Атланта, США). Програма «MILE» навчає дитину зосереджуватись та міркувати за допомогою стратегії УРА: «Увага-Робота-Аналіз». Цей тип фокусування та розмірковування допомагає дитині пов‘язувати дії та явища і сприяє розвитку пізнавальної здатності. Програма призначена для занять з дитиною у супроводі корекційних педагогів, дефектологів, психологів та проведення домашніх занять. Програма «MILE» перекладена з англійської і адаптована українською мовою за підтримки МБФ «ОМНІ-мережа для дітей».
Ще однією важливою проблемою розвитку та навчання дітей із ФАС є засвоєння ними поведінкових та побутово – адаптивних навичок, оскільки вплив алкоголю спричиняє проблеми щодо уваги та регуляції збудження. На допомогу таким дітям розроблена науковцями зі США (університету Еморі , Атланта) і надана для адаптації в Україні програма з робочою назвою «Go FAR» (як спектр застосування стратегії УРА). Завдання програми – розвинути у дітей навички соціальної взаємодії, побутові вміння (так звані, адаптивні навички). Програма призначена і для батьків, і для дітей. Цікавим додатком до програми є комп‘ютерна гра на подолання імпульсивності та розвитку здатності планувати свої дії.
Зважаючи на реалії сьогодення, дуже важливо, щоб у житті дітей, які зазнали пренатального впливу алкоголю, були присутні вісім універсальних захисних факторів. Захисний фактор – це будь-що, що не дає одній проблемі спровокувати розвиток іншої. До цих захисних факторів належать:
- Діагноз у віці до 6 років;
- Встановлений діагноз ФАС;
- Забезпечення основних потреб;
- Проживання у стабільному місці понад 72% усього життя;
- Ніколи не переживати насильства стосовно себе;
- Проживання у кожному місці в середньому більше, ніж 2,8 роки;
- Проживання в хороших умовах з 8-12 років;
- Отримання допомоги від спеціалістів з розвитку.
А послання до суспільства є простим і зрозумілим, не тільки 9 вересня, але кожного дня:
ЖІНКА, ЯКА Є АБО МОЖЕ БУТИ ВАГІТНОЮ, АБО ПЛАНУЄ ВАГІТНІСТЬ, ПОВИННА КАТЕГОРИЧНО ВІДМОВИТИСЬ ВІД ВЖИВАННЯ АЛКОГОЛЮ!
Переглянуто редакційною колегією I.B.I.S.: 03/09/2016
Дивіться також:
- Алкоголь та здоров’я (інформація для батьків)
- Алкогольний синдром плода (інформація для батьків)
- Вживання алкоголю під час вагітності (інформація для батьків)
- Фетальний алкогольний синдром
Целіакія
(Celiac Disease)
Т.О. Головатюк
Спеціаліст з інформаційного забезпечення
Хмельницького ОМНІ-Центру
Целіакія – одне із найбільш поширених генетичних захворювань, що вражає тонкий кишечник і приводить до порушення всмоктування поживних речовин з їжі. Люди з діагнозом целіакії не переносять білок глютен, який є у пшениці, житі, ячмені і, можливо, вівсі. Коли хворі на целіакію вживають продукти, що містять глютен, їх імунна система відповідає на це пошкодженням тонкого кишечника. На внутрішній його поверхні відмирають крихітні пальцеподібні ворсинки, так звані “віллі”, які виконують у кишечнику важливу роль: через них поживні речовини з їжі всмоктуються в кров. Без цих ворсинок організм виснажується незалежно від кількості вживаної їжі. Поверхня кишечника пошкоджується місцями. На внутрішній поверхні тонкого кишечника одночасно можуть знаходитися одна чи кілька “мертвих” плям.
Целіакію розглядають як аутоімунний розлад, оскільки власна імунна система викликає пошкодження. ЇЇ також класифікують і як “порушене засвоювання”, так як поживні речовини не всмоктуються в кров.
Целіакія – найчастіше генетичне захворювання у Європі. В Італії приблизно 1 людина із 250, в Ірландії – 1 з 300 страждають целіакією. У осіб китайського, японського і африканського походження хвороба виявляється рідко. В США целіакія виявляється з частотою 1 на 4700 мешканців.
Які симптоми хвороби?
Прояви целіакії різноманітні, як і час дебюту захворювання (дитинство, дорослий вік). Одним із факторів, від яких залежить клінічна картина целіакії, є грудне вигодовування: чим довше мати годує дитину грудьми, тим пізніше може проявитися захворювання і тим більш нетиповими і маловиразними можуть бути симптоми. Іншими факторами є вік, в якому дитину почали годувати продуктами, що містять глютен, і вміст глютену в них.
Основними симптомами хвороби є розлади травлення. Але захворювання може проявлятися і по-іншому. Наприклад, в одного хворого можуть з’явитися діарея і болі в черевній порожнині, тоді як у іншого – депресія або надмірна збудженість. Надмірна дратівливість – також один із характерних симптомів у дітей.
Симптоми захворювання:
- відчуття здуття та болю в черевній порожнині, що має постійний характер;
- хронічна діарея;
- втрата у вазі;
- слабо забарвлений стілець з неприємним гнилісним запахом;
- анемія (мала кількість червоних тілець у крові);
- м’язові судоми;
- хронічна втома;
- відставання у рості;
- відхилення розвитку у дітей;
- біль у суглобах;
- поколювання та оніміння в ногах (через пошкодження нерва);
- бліді виразки в роті (aphtus ulcers);
- хвороблива висипка на шкірі (dermatitis herpetiformіs);
- декальцинація зубів або стирання емалі;
- збої в менструальному циклі (часто через надмірну втрату у вазі);
- безпліддя як у жінок, так і в чоловіків.
Анемія, відставання в рості і втрата ваги – це ознаки виснаження, дефіциту поживних речовин в організмі. Виснаження – серйозна проблема для усіх, але особливо для дітей, яким для повноцінного розвитку необхідна певна кількість поживних речовин.
У деяких людей може не бути жодних очевидних симптомів. Це зв’язано з тим, що неушкоджена частина їх тонкого кишечника всмоктуючи достатню для організму кількість поживних речовин, маскує ураження інших ділянок. Однак у хворих, які тимчасово не мають симптомів целіакії, ризик виникнення її ускладнень не менший.
Як діагностувати целіакію?
Целіакія – хвороба, яку важко діагностувати. Це пояснюється неспецифічністю симптомів, їх схожістю з іншими захворюваннями, наприклад, синдромом подразнення кишківника, хворобою Крона, виразковим колітом, кишковими інфекціями, синдромом хронічної втоми та депресії. Нещодавно дослідники виявили, що хворі на целіакію мають підвищений вміст у крові певних антитіл: антигліадину, антиендомізиуму і антиретикуліну. Вони виробляються імунною системою у відповідь на чинники, які організм сприймає як загрозливі для здоров’я подразники.
Для діагностики целіакії лікарі досліджують кров і визначають рівень антитіл до глютену. Якщо результати тесту і симптоми вказують на целіакію, лікар може взяти крихітну частину тканини тонкої кишки, щоб з’ясувати , чи не пошкоджені ворсинки. Це робиться під час процедури, яка називається біопсією тонкого кишечника: вводять довгу тонку трубку (ендоскоп) через рот і шлунок в тонку кишку і потім беруть потрібний зразок тканини за допомогою інструмента, пропущеного через ендоскоп.
Які ускладнення целіакії?
Пошкодження тонкої кишки при целіакії і, як наслідок, порушення засвоєння, поживних речовин підвищують ризик виникнення деяких хвороб та інших проблем зі здоров’ям:
- лімфома і аденокарцинома – ці типи раку можуть виникнути в кишечнику;
- остеопороз – крихкість кісток, що підвищує ризик їх переломів. Погане засвоєння кальцію – фактор, який сприяє остеопорозу;
- порушення внутрішньоутробного розвитку (дефекти нервової трубки) у дітей жінок, які під час вагітності мали порушення засвоєння поживних речовин;
- низькорослість, якщо хвороба проявилась у дитячому чи підлітковому віці та не була адекватно пролікована.
Оскільки целіакія є аутоімунною хворобою, то у хворих на неї осіб можуть виникати і інші аутоімунні розлади, як от:
- герпетиформний дерматит;
- аутоімунний тиреоїдит;
- системний червоний вовчак;
- інсулінзалежний діабет;
- аутоімунний гепатит;
- аутоімунні захворювання судин (васкуліти);
- ревматоїдний артрит;
- синдром С’єгрена (загальна сухість слизових оболонок).
Причини помилкової та запізнілої діагностики целіакії:
- симптоми целіакії приписуються іншим хворобам;
- багато лікарів недостатньо інформовані відносно цієї хвороби;
- є дуже мало лабораторій, де можуть якісно зробити відповідні аналізи.
Чи існує профілактика целіакії?
З метою ранньої діагностики рекомендується проведення тестування – визначення антитіл до глютену людям без симптомів захворювання. Оскільки целіакія – захворювання спадкове, повинні бути перевірені члени родини хворого, особливо найближчі родичі. В 10% випадків целіакія виявляється і у них. Слід мати на увазі, що чим довше не виявляється і не лікується це захворювання, тим вища ймовірність розвитку виснаження організму та інших незворотних ускладнень.
Наприклад, в Італії, де целіакія – захворювання дуже поширене, проводиться профілактичне обстеження усіх дітей у віці 6 років. Завдяки такій пильності італійців з моменту появи симптомів до моменту встановлення діагнозу проходить звичайно 2-3 тижні. В США – біля 10 років.
Як лікувати целіакію?
Єдиним ефективним методом лікування целіакії сьогодні є безглютенова дієта – повне виключення усіх продуктів, що містять глютен. Вона допомагає поступово відновити пошкоджену частину кишечника, захистити його від нового пошкодження і позбавитись від важких симптомів. Покращення настає з перших же днів застосування дієти, а повноцінно ворсинки починають працювати через 3-6 місяців у дітей і майже через 2 роки у дорослих.
Дотримання безглютенової дієти – пожиттєва вимога до хворого, тому що навіть найменша кількість глютену може викликати нові пошкодження кишечника. Ця вимога однакова для усіх хворих на целіакію, включаючи і тих, у кого немає очевидних симптомів. Вважається, що залежно від віку, в якому встановлений діагноз, від деяких проблем, таких як відставання в рості і декальцинація зубів, позбавитись вже неможливо.
Варто знати, що деяким хворим на целіакію безглютенова дієта не допомагає: у них настільки пошкоджений кишечник, що вже не в змозі відновитися навіть після виключення з раціону продуктів з вмістом глютену. Ці люди можуть потребувати внутрішньовенного введення поживного розчину, а їх стан розцінюється як ускладнення захворювання.
Якщо організм пацієнта реагує на безглютенову дієту, лікар може підтвердити, що діагноз целіакії встановлений правильно.
Що таке безглютенова дієта?
Вона означає виключення з раціону всіх продуктів, які містять пшеницю, жито, ячмінь і, можливо, овес, а це значить, хліб, макарони, каші і багато інших похідних продуктів. Однак хворі на целіакію можуть харчуватися при добре збалансованій дієті, яка включає хліб і макарони із спеціального борошна. Наприклад, замість пшеничного можна використовувати картопляне, рисове або бобове. Безглютенові продукти можна купувати в спеціалізованих магазинах.
Безглютенова дієта складна. Вона вимагає абсолютно нового підходу до харчування, що ускладнює життя людини. Хворі діти повинні бути дуже обережні, купуючи обід в школі, дорослі – при виборі закусок на званих вечірках і т.д. Прийом їжі – іспит для хворого целіакією: мусиш ретельно вивчати меню і випитувати у офіціанта точний склад блюда, щоб уникнути глютену. Тим не менше, така пильність стає другою натурою, і людина вчиться розпізнавати допустиму і недопустиму для неї їжу.
Перелік основних безпечних і небезпечних продуктів
для хворих на целіакію
Переглянуто редакційною колегією I.B.I.S.: 24/06/2003
Хвороба Фaбpi
(Fabry Disease)
Галина Петрівна Оскрес
Лікар-генетик
Бережанської центральної районної лікарні Тернопільської області
Синоніми:
- Дефіцит α-галактозидази А.
- Хвороба Андерсона-Фабрі.
- Дифузна універсальна ангіокератома.
- Спадковий дистопічний ліпідоз.
- Порушення обміну глікосфінголіпідів.
Етіологія та патогенез:
Причиною розвитку клінічної картини є мутація в гені лізосомального ферменту α-галактозидази А (α-GLA), який картовано на хромосомі Х, сегмент q-22.1, що призводить до повної відсутності або зменшення активності ферменту, що приймає участь у метаболізмі ліпідів з кінцевими α-галактозними залишками, особливо глоботріазілцераміду (ГЛ-З або ГБ-3), які накопичуються в лізосомах клітин організму (ендотелій, гладкі м’язи ниркової та серцево-судинної систем, ретикулоендотелій, сполучна тканина, рогівка, міокард). Основна симптоматика хвороби Фабрі обумовлена ішемією та мікроінфарктом в уражених тканинах внаслідок прогресуючого накопичення глікосфінголіпідів. Дотепер описано більш ніж 200 різних мутацій в гені α-GLA.
Основні діагностичні критерії:
- Акропарестезії.
- Шкірні прояви у вигляді ангіокератоми.
- Помутніння рогівки i кришталика.
- Рецидивуючі підвищення температури тіла в дитинстві, асоційовані з болями в кінцівках.
- Гіпогідроз або ангідроз.
- Інсульт, гіпертрофія лівого шлуночка.
- Протеїнурія та ниркова недостатність невідомої етіології.
- 3ниження активності α-галактозидази А в плазмі крові, в лейкоцитах, в культурі фібробластів.
- Виявлення мутації гену α-GLA на X-q22 при молекулярно генетичному дослідженні.
Клініка:
Розрізняють найбільш важку класичну форму хвороби Фабрі, а також нирковий та серцевий варіанти.
Початок захворювання спостерігається зазвичай у дитинстві або у підлітковому віці. До ранніх симптомів відносять: періодичні парестезії та акропарестезії, ангіокератоми, помутніння рогівки ока та кришталика, гіпогідроз, порушення функції травлення (болі в животі, діарея, блювання), непереносимість фізичних та емоційних навантажень, зміни аналізу сечі у вигляді протеїнурії та ізостенурії. З віком (підлітковий та дорослий вік) приєднуються порушення функції нирок, серцево-судинні захворювання (інфаркт, гіпертрофія серця, вади клапанів, аритмії), цереброваскулярні ускладнення (інсульт, транзиторні ішемічні атаки), порушення зовнішнього дихання, а також втрата слуху, запаморочення.
Парестезії можуть тривати від декількох хвилин до декількох днів та супроводжуватися підвищенням температури тіла, підвищенням ШОЕ. Болі зазвичай провокуються фізичними та емоційними перевантаженнями, швидкою зміною температури та вологості. Біль може віддавати в інші ділянки тіла, так, приступ болю в животі може симулювати апендицит або ниркову кольку. 3 віком у більшості хворих приступи частішають, стають більш інтенсивними та тривалими, вимагаючи для їх купування не лише карбамазепіну чи фенітоїну, а часом і наркотичних анальгетиків.
Ангіокератоми є однією з перших ознак хвороби Фабрі, проте вони не є патогномонічними. З’являються ангіокератоми як скупчення темно-червоних та темно синіх ангіоектазій в поверхневих шарах шкіри. Найбільш виражені вони в ділянці між пупком та колінами, тобто на спині, сідницях, ділянці статевих органів. Кількість уражених ділянок шкіри збільшується з віком.
Ураження кришталика, так звана “катаракта Фабрі” (білясті плямисті відкладення поблизу задньої капсули кришталика), зустрічається у 30% чоловіків. Помутніння рогівки при спеціальному офтальмологічному дослідженні виявляється у більшості випадків не тільки у хворих, але і у гетерозиготних жінок. Але ураження рогівки може бути відсутнім при кардіальному варіанті хвороби.
Прогресуюче накопичення глікосфінголіпідів у нирках призводить до порушення канальцевої реабсорбції, секреції та екскреції, внаслідок чого наростає азотемія та інші ознаки ниркової недостатності: протеїнурія, поліурія, ізостенурія, уремія. Заключна стадія ниркової недостатності розвивається у третьому-п’ятому десятиріччі життя і є однією з основних причин смерті пацієнтів з хворобою Фабрі.
Зміни з боку серцево-судинної системи з’являються переважно в другій половині життя. Мітральна недостатність може спостерігатись ще з дитячого віку. Накопичення ліпідів в міокарді веде до гіпертрофії лівого шлуночка та інфільтрації провідної системи серця з появою аритмій. До пізніх симптомів відносять гіпертензію, ішемію міокарда та інфаркт, застійну серцеву недостатність, мітральну регургітацію.
Причиною цереброваскулярних порушень, що у випадку класичного варіанту хвороби Фабрі дебютують на 4-му десятиріччі життя, є інфільтрація малих судин, що проявляється тромбозами, транзиторними ішемічними атаками, судомами, геміплегією та ін.
Накопичення глікосфінголіпідів призводить i до ураження інших органів: шлунково-кишкового тракту (діарея, нудота, болі в животі), кісткової системи (деформації дистальних міжфалангових суглобів, остеопороз хребців, асептичний некроз головки стегнової кістки), гіпохромної мікроцитарної анемії.
Кардіальний варіант хвороби спостерігається при наявності остаточної активності ферменту α-галактозидази А. Перші прояви хвороби у вигляді гіпертрофії лівого шлуночка, мітральної недостатності, кардіоміопатії, протеїнурії з нормальною функцією нирок з’являються на шостому-восьмому десятиріччі життя. Типовими є синусова брадікардія, зміни ЕКГ, як от: інверсія зубця Т, вкорочення PR інтервалу, неспецифічні зміни сегменту ST. Ниркова недостатність, зазвичай, не розвивається. Більшість пацієнтів спостерігаються як хворі з кардіоміопатією та залишаються не діагностованими.
Нирковий варіант хвороби Фабрі було описано нещодавно у 6 хворих із нирковою недостатністю, які мали помилковий діагноз хронічного гломерулонефриту. П’ять з них не мали шкірних проявів, парестезій, гіпогідрозу та помутніння рогівки, але мали гіпертрофію лівого шлуночка. У всіх хворих було виявлено зниження активності α-галактозидази в плазмі крові, а проведене молекулярно-генетичне дослідження виявило специфічну міссенс-мутацію гену GLA.
Клінічні прояви у гетерозиготних носіїв демонструють значну варіабельність – від асимптоматичного перебігу до помірної вираженості проявів, рідко з розвитком важкої форми, як у чоловіків. Такі відмінності перебігу виникають внаслідок випадкової інактивації однієї з Х хромосом у жінок-носіїв під час раннього ембріогенезу. Зазвичай клініка набагато легша порівняно з хворими чоловіками. Клінічні ознаки поділяються на легкі, до яких відносять помутніння рогівки та кришталика (70-90%), що не порушує зорову функцію, ізольовані ангіокератоми (10-50%), акропарестезії (50-90%), та важкі, що загрожують життю: значна гіпертрофія міокарда, ішемія міокарда та інфаркт, аритмії, транзиторні ішемічні атаки, інсульт. Ниркові ураження у гетерозиготних жінок можуть проявлятись ізостенурією, гематурією, лейкоцитурією, присутністю гранулярних та гіалінових циліндрів. Однак лише у 10% носіїв може розвинутися ниркова недостатність, що потребує гемодіалізу та пересадки нирок.
Ускладнення:
- Прогресуюча ниркова недостатність, що призводить до термінальної стадії ураження нирок, виникає в середньому після 25 років.
- Серцево-судинна дисфункція: гіпертрофія лівого шлуночка, інфаркт міокарда.
- Судинно-мозкові ускладнення: транзиторні ішемічні атаки, збільшується ризик ранніх інсультів.
- Легеневі ускладнення: закупорка дихальних шляхів, диспное.
Диференційний діагноз:
Хворобу Фабрі, перш за все, необхідно диференціювати зі спадковою геморагічною телеангіоектазією, для якої не характерні приступи болю – акропарестезії; з ревматичним i ювенільним ревматоїдним артритом, при яких основними проявами є зміни з боку суглобів, ураження дистальних міжфалангових суглобів, ранкова скутість, запальні зміни в крові; з іншими формами ангіокератом, що потребує гістологічного дослідження; з гіпертрофічною кардіоміопатією. Також необхідно виключати інші лізосомні хвороби накопичення: фукозидоз, сіалідоз (дефіцит α-нейрамінідази з або без дефіциту β-галактозидази), аспартатглюкозамінурію, дефіцит β-маннозидази, дорослий тип дефіциту α-галактозидази В.
Співвідношення статей:
Ч 1 : Ж 0. Хворіють чоловіки. Жінки є гетерозиготними носіями.
Частота: 1:50000 чоловіків.
Тип успадкування:
Х-зчеплений рецесивний. Ризик для сибсів чоловічої статі складає 50%, для сибсів жіночої статі 50% бути носієм. Ризик для доньок хворого чоловіка – 100% бути носієм, сини не успадковують мутантний ген і тому є здоровими. Ризик для нащадків гетерозиготної жінки успадкувати мутантний ген становить 50%, нащадки чоловічої статі, що успадкували мутантний ген хворітимуть синдромом Фабрі, нащадки жіночої статі будуть гетерозиготними носіями.
Єдиним методом виявлення гетерозиготного носійства є молекулярно-генетичне дослідження.
Лікування:
Тривалий час хворобу Фабрі можна було лікувати тільки симптоматично: карбамазепін i діфенілгідантіон при акропарестезіях; лазерне лікування ангіокератом; встановлення штучного водія ритму; операція аорто-коронарного шунтування при ішемії міокарда; гемодіаліз та пересадка нирки при термінальній стадії ниркової недостатності; бронходилятаційна терапія та ін.
На сьогоднішній день розроблена методика ензимо-замісної терапії хвороби Фабрі. Застосовується Фабразім (algalsidase beta, Genzyme Corp) та Реплагал (algalsidase alpha, Transkaryotic Therapies, Inc) – рекомбінантні форми людської α-галактозидази А. Рекомендується розпочинати ензимо-замісну терапію одразу після встановлення діагнозу, включаючи хворих із термінальною стадією ниркової недостатності та жінок гетерозиготних носіїв, що мають клінічні прояви хвороби. Ранній початок замісної терапії може полегшити, або навіть попередити розвиток багатьох симптомів хвороби.
Профілактика:
Пренатальна діагностика на 10-12 тижнях вагітності з визначенням статі плоду, та визначенням активності α-галактозидази А в фетальних клітинах плода чоловічої статі, а також проведення молекулярно генетичної діагностики при відомій мутації в гені GLA в родині.
Номер з каталогу МІМ:
301500 Fabry Disease.
Література:
- Baehner F, Kampmann C, Whybra C, Miebach E, Wiethoff CM, Beck M . Enzyme replacement therapy in heterozygous females with Fabry disease: results of a phase IIIB study. J Inherit Metab Dis 2003;26:617-27 [Medline].
- Bennett RL, Hart KA, O’Rourke E, Barranger JA, Johnson J, MacDermot KD, Pastores GM, Steiner RD, Thadhani R. Fabry disease in genetic counseling practice: recommendations of the National Society of Genetic Counselors. J Genet Couns 2002;11:121-46 [Medline].
- Desnick RJ. Enzyme replacement therapy for Fabry disease: lessons from two alpha-galactosidase A orphan products and one FDA approval. Expert Opin Biol Ther 2004;4:1167-76 [Medline].
- Desnick RJ, Banikazemi M, Astrin KH. Genetics of Fabry Disease (https://emedicine.medscape.com/).
- Desnick RJ, Brady R, Barranger J, Collins AJ, Germain DP, Goldman M, Grabowski G, Packman S, Wilcox WR. Fabry disease, an under-recognized multisystemic disorder: expert recommendations for diagnosis, management, and enzyme replacement therapy. Ann Intern Med 2003;138:338-46 [Medline].
- Galanos J, Nicholls K, Grigg L, Kiers L, Crawford A, Becker G. Clinical features of Fabry’s disease in Australian patients. Intern Med J 2002;32:575-84 [Medline].
- MacDermot KD, Holmes A, Miners AH. Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. J Med Genet 2001;38:769-75 [Medline].
- Nakao S, Kodama C, Takenaka T, Tanaka A, Yasumoto Y, Yoshida A, Kanzaki T, Enriquez AL, Eng CM, Tanaka H, Tei C, Desnick RJ. Fabry disease: detection of undiagnosed hemodialysis patients and identification of a “renal variant” phenotype. Kidney Int 2003;64:801-7 [Medline].
- Nakao S, Takenaka T, Maeda M, Kodama C, Tanaka A, Tahara M, Yoshida A, Kuriyama M, Hayashibe H, Sakuraba H, et al. An atypical variant of Fabry’s disease in men with left ventricular hypertrophy. N Engl J Med 1995;333:288-93 [Medline].
- Whybra C, Kampmann C, Willers I, Davies J, Winchester B, Kriegsmann J, Bruhl K, Gal A, Bunge S, Beck M. Anderson-Fabry disease: clinical manifestations of disease in female heterozygotes. J Inherit Metab Dis 2001;24:715-24 [Medline].
Переглянуто редакційною колегією I.B.I.S.: 25/05/2005
Трисомія 18 – обвід голови
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”
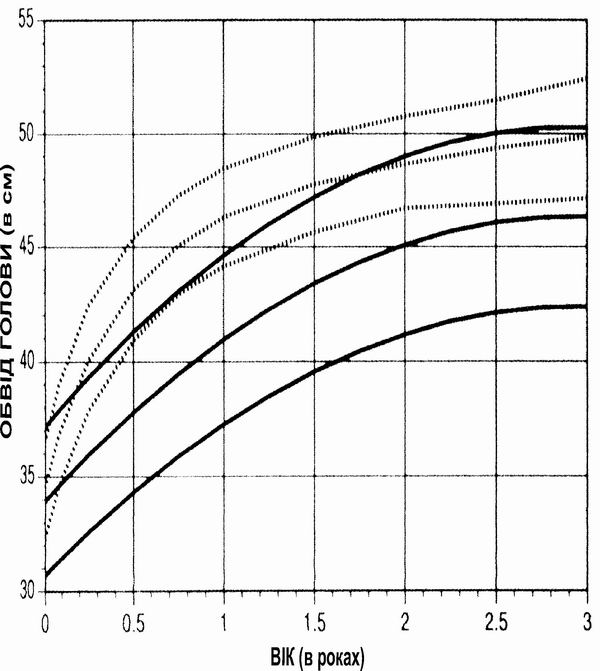
Криві збільшення обводу голови в пацієнтів з Трисомією 18 від народження до 18-річного віку (суцільні лінії) порівнюються з даними розвитку здорових осіб (пунктирні лінії). Вимірювання (84% – дівчатка) проводились у 76 пацієнтів. Baty BJ et al. Natural History of Trisomy 18 and Trisomy 13:I. Growth, Physical Assessment, Medical Histories, Survival, and Recurrence Risk. Am J Med Genet 49:175-188 (1994) Copyright (c) 1994 Wiley-Liss, Inc. Reprinted by permission of Wiley-Liss, Inc., a division of John Wiley & Sons, Inc.
Тетрада Фалло
(Tetralogy of Fallot)
Т.Є. Голуб
Відділення патології новонароджених
Рівненської обласної дитячої лікарні
Синонім:
Атрезія устя легеневої артерії з дефектом міжшлуночкової перетинки.
Включення:
Включає 4 анатомічні ознаки:
- Дефект міжшлуночкової перетинки (ДМШП).
- Інфундибулярний стеноз легеневої артерії.
- Декстрапозиція аорти, що локалізується над ДМШП.
- Гіпертрофія правого шлуночка (ПШ).
Анатомія:
Стеноз легеневої артерії при тетраді Фалло обов’язково інфундибулярний (низький, високий або у вигляді дифузної гіпоплазії). Часто він поєднується з клапанним стенозом, гіпоплазією кільця (60%) і стовбура легеневої артерії (близько 70%). При поєднанні клапанного і інфундибулярного стенозів вивідний тракт між ними може розширюватись, утворюючи вихідну кишеню, так званий “третій шлуночок”. З віком ступінь стенозу вивідного тракту (фібром’язовий утвір) зростає, тобто вада має прогресуючий перебіг. Крайню форма обструкції являє собою атрезія клапанів легеневої артерії (20%). Нерідко зустрічається гіпоплазія та стеноз гілок легеневої артерії, можлива атрезія лівої гілки. ВАП (відкрита артеріальна протока) функціонує, як правило, у дітей перших місяців життя і її закриття веде до погіршення стану хворого. При атрезії клапанів легеневої артерії легеневий кровоток здійснюється за рахунок ВАП або аортолегеневих коллатеральних судин.
ДМШП при тетраді Фалло завжди великий, розміщений нижче надшлуночкового гребеня під коренем аорти.
Гіпертрофія міокарду правого шлуночка поєднується з нормальними за розмірами лівими відділами серця. При різко вираженому легеневому стенозі порожнина лівого шлуночка може бути зменшена.
При тетраді Фалло мають місце виражені морфологічні зміни судин малого кола кровообігу. На початковій стадії вони незначні і полягають у розростанні інтими. Потім периферичні судини значно звужуються, з’являються ангіоматозні структури, артеріовенозні анастомози. В 10-15 річному віці і пізніше з’являється схильність до тромбоутворення в легеневих судинах. Такими змінами пояснюються несприятливі результати операції.
Асоційовані вади:
Зустрічаються в 40% випадків: правобічна дуга аорти (20-25% частіше при гіпоплазії або атрезії легеневої артерії); судинне кільце, вторинний ДМПП, ВАП, відкритий загальний атріовентрикулярний канал, додаткова лівобічна верхня порожниста вена, частковий аномальний дренаж легеневих вен.
Клініко-анатомічні варіанти тетради Фалло:
- Тетрада Фалло з атрезією устя легеневої артерії.
- Класична форма з різноманітним ступенем стенозу.
- Бліда форма тетради Фалло.
Гемодинамічні зміни:
Тетрада Фалло відноситься до вад, що викликають гемодинамічні зміни у плода. Тиск в системному і легеневому колах кровообігу у плода підтримується на однаковому рівні. Навіть при вираженому стенозі або атрезії легеневої артерії кров з правого шлуночка надходить в аорту і легеневий кровообіг здійснюється шляхом ретроградного кровотоку через артеріальну протоку. Це підтверджується нормальним внутрішньоутробним ростом плода з тетрадою Фалло і відсутністю гіпертрофії правого шлуночка у новонародженого.
Тетрада Фалло з відсутністю клапана легеневої артерії відноситься до виключення, оскільки легенева регургітація призводить до застійної серцевої недостатності. У такому випадку у дитини порушується функція дихання внаслідок зовнішнього стиснення бронхів і трахеї аневризматично розширеною основною легеневою артерією та її гілками.
Після народження гемодинамічні порушення пов’язані з шунтуванням крові зліва направо на рівні висхідної аорти і обхідного легеневого кровообігу. Зменшення насичення системної крові киснем супроводжується ціанозом.
Поєднання інфундибулярного стенозу легеневої артерії і системного тиску в аорті веде до перевантаження і гіпертрофії правого шлуночка.
У випадках помірного стенозу, коли опір викиду крові в легені нижчий, ніж в аорті, наявний ліво – правий скид, що клінічно проявляється блідою (аціанотичною) формою тетради Фалло. Вона частіше зустрічається у дітей перших років життя і дорослих. При зростанні стенозу виникає перехресний, а потім стабільно право-лівий (вено – артеріальний) скид крові. Клінічно це означає перехід в ціанотичну форму вади.
Ліві відділи серця при тетраді Фалло функціонально недовантажені, що є причиною відносної гіпоплазії лівого шлуночка. У дорослих хворих з вираженим колатеральним кровообігом лівий шлуночок має достатні розміри.
Пренатальна діагностика:
Тетраду Фалло діагностують на основі виявлення розширення аорти, що поширюється на міжшлуночкову перегородку. На ранніх термінах гестації інфундибулярний стеноз легеневої артерії може бути невираженим.
При зміщенні аорти, коли не визначається відходження легеневої артерії від правого шлуночка, слід проводити диференційну діагностику з атрезією легеневої артерії, з ДМШП, з загальним артеріальним стовбуром. При з’єднанні аорти з легеневими артеріями діагностують артеріальний стовбур. Аневризматичне розширення легеневої артерії свідчить про відсутність її клапана.
Диференційний діагноз:
Клінічно слід проводити з транспозицією аорти і легеневої артерії, подвійним відходженням магістральних судин від правого шлуночка, єдиним шлуночком, двокамерним серцем зі стенозом легеневої артерії.
Етіологія:
Можна виділити такі причини виникнення тетради Фалло:
- Хромосомні аберації – 5%.
- Мутація одного гена – 2-3%. Такі вади також поєднуються з аномаліями розвитку інших органів.
- Фактори навколишнього середовища (алкоголізм батьків, краснуха, ліки і ін.) – 1-2%. Так, у жінок з ревматизмом у 25% випадків народжувалися діти з ВВС; частота ВВС при алкоголізмі у матері в 3 рази більша від звичайної; у жінок, що перенесли краснуху в І-ому триместрі вагітності, 70-80% дітей мають ВВР.
- Полігенно-мультифакторіальне успадкування – 90%.
Доведено тератогенну дію з розвитком даної вади при вживанні матір’ю дитини триметадіону та прогестагенів.
Найбільша вірогідність виникнення ВВС на 8-12-ому тижні гестації, коли відбувається ембріональна закладка і формування серцево-судинної системи.
Для тетради Фалло тератогенна дія патологічних факторів починається з 18-го дня гестації і закінчується до 29-го дня.
Факторами ризику народження дитини з ВВС є вік матері 15-17 років; більше 40 років; ендокринні порушення (цукровий діабет); токсикоз в І-ому триместрі вагітності; загроза переривання вагітності; мертвонароджуваність в анамнезі; наявність інших дітей з ВВР; прийом жінкою гормональних препаратів для збереження вагітності; діти малих термінів гестації.
Частота виникнення:
ВВС серед всіх ВВР складають 10%, зустрічаються з частотою 8 випадків на 1000 живонароджених. Частота тетради Фалло – 1 випадок на 5000 живонароджених.
Клінічна картина, діагностика:
Найбільш важкі форми виявляються в перші дні життя. Діагноз встановлюють при наявності грубого систолічного шуму тахіпное, ціанозу. Ціаноз – один з головних симптомів тетради Фалло. Час його появи та вираженість визначають ступінь стенозу легеневої артерії. З народження він спостерігається в 40% випадків, частіше стає вираженим в 6-12 місяців по мірі зростання активності дитини. Симптом “барабанних паличок” та “годинникових скелець” з’являється в залежності від ступеня гіпоксемії на 1-2-ому році життя. Толерантність до фізичного навантаження у таких дітей знижена.
Основним симптомом, що виражає важкість стану і розвиток ускладнень з боку ЦНС, є ціанотично-ядухові напади. Вони відсутні у дітей перших 3-х місяців життя і виникають, перебігаючи найбільш важко у віці від 6 до 24 місяців. Виникнення нападів пов’язане зі спазмом інфундибулярного відділу правого шлуночка, внаслідок чого вся венозна кров через ДМШП надходить в аорту і викликає або посилює гіпоксію ЦНС.
При поєднанні тетради Фалло з судинним кільцем відмічається стрідорозне дихання та ознаки стиснення стравоходу і трахеї.
Гіпотрофія ІІ-ІІІ ступеня характерна в 36,7% випадків. У більш старшому віці і у дорослих характерне відставання у фізичному розвитку. У дітей 1-го і 2-го років життя відмічається різкий ціаноз і затримка моторного розвитку. Для хворих з тетрадою Фалло характерні повторні гострі респіраторні інфекції, карієс, хронічний тонзиліт, гайморит.
При тетраді Фалло ядуха по типу диспное – поглиблене аритмічне дихання. Серцевий горб відсутній. Іноді відмічається систолічне тремтіння. При аускультації – звучний І-й тон; грубий систолічний шум з максимумом в ІІI і IV міжребер’ях зліва (при інфундибулярному стенозі) і в ІІ міжребер’ї (при поєднанні з клапанним). Другий тон ослаблений над легеневою артерією.
Серцева недостатність по правошлуночковому типу не характерна для тетради Фалло, а тахікардія і ядуха пов’язані з гіпоксією. Поява серцевої недостатності при тетраді Фалло у дорослих хворих прогностично несприятлива ознака, що вказує на різкий стеноз і поширений кардіосклероз.
З народження у дітей з важкою формою тетради Фалло відмічається різкий ціаноз, ядуха, тахікардія (що не піддається медикаментозній корекції), зниження маси тіла, гіподинамія, гіпоксичні приступи, рано деформуються нігтьові фаланги, можливі прояви правошлуночкової серцевої недостатності. При аускультації систолічний шум не прослуховується, може прослуховуватись систолодіастолічний шум в ІІ-ому міжребер’ї зліва (ВАП), або на спині (шум коллатералей).
У хворих раннього віку з тетрадою Фалло можна виділити 3 клінічні фази вади (Білоконь Н.А. 1971):
- Фаза відносного клінічного благополуччя (з народження до 6 місяців), нема відставання в фізичному розвитку, стан майже задовільний, напади частіше відсутні.
- Фаза ядухово-ціаностичних нападів (від 6 місяців до 24 місяців), має місце велика кількість ускладнень з боку ЦНС і летальних випадків, так як відбувається вікова перебудова еритропоезу та становлення інших компенсаторних механізмів, одночасно збільшується гемодинамічний ефект стенозу.
- Перехідна фаза, коли клінічна картина вади починає набувати риси, характерні для дітей старшого віку, незважаючи на наростання ціанозу, зникають напади (або попереджуються, якщо дитина займає вимушене положення – навприсядки), зменшується ядуха і тахікардія, в крові має місце поліцетимія і поліглобулія, в легенях – коллатеральний кровообіг.
Лабораторні та інструментальні методи діагностики:
ЕКГ: спостерігаються відхилення електричної осі серця вправо, ознаки гіпертрофії правого шлуночка. Неповна блокада правої ніжки пучка Гіса має місце у 1/5 хворих. Часто зустрічаються порушення ритму (екстрасистолія), ознаки гіпертрофії правого передсердя.
ФКГ: реєструється систолічний шум ромбоподібної форми з максимумом в першу половину систоли, легеневий компонент ІІ-го тону значно ослаблений або відсутній (при вираженому стенозі).
Рентгенограма органів грудної клітини: легеневий малюнок збіднений, рідше посилений за рахунок коллатералей. Типова форма серця у вигляді “чобітка” за рахунок заокругленої, припіднятої над діафрагмою верхівки і западання дуги легеневої артерії, тінь серця невелика. Має місце гіпертрофія правого шлуночка, лівий шлуночок невеликий; по лівому контуру серця спостерігається випинання (“третій шлуночок”), можливе збільшення правого передсердя.
М-сканограма: відсутність взаємного переходу передньої стінки аорти в міжшлуночкову перетинку, при цьому широка аорта розміщена над дефектом. Вторинні ознаки: потовщення стінки правого шлуночка і міжшлуночкової перегородки, зменшення лівого передсердя, розширення кореня аорти.
Двомірна ехокардіографія: дозволяє визначити величину зміщення аорти, ДМШП, ступінь легеневого стенозу і гіпертрофію ПШ.
Катетеризація порожнин серця: визначається високий тиск в ПШ, рівний системному градієнту тиску між правим шлуночком і легеневою артерією. Низький тиск в останній відображає ступінь стенозу. Тиск в ПШ частіше нормальний. Насичення артеріальної крові киснем складає в середньому 70%, при атрезії ЛА ще менше. Проходження катетера з правого шлуночка в аорту є характерною ознакою тетради Фалло.
Рентгенконтрастне дослідження: легенева артерія заповнюється контрастом одночасно. При атрезії легеневої артерії визначають відсутність надходження контрастної речовини з правого шлуночка в легеневу артерію, і після контрастування дуги і низхідного відділу аорти видно слабе контрастування внутрішньолегеневих розгалужень або гілок легеневих артерій (через колатеральні судини або ВАП). Аортографія і легенева артеріографія виявляють колатеральний кровообіг, ВАП, патологію стовбура легеневої артерії та її гілок.
Загальний аналіз крові: абсолютна (гемоглобін менше 107 г/л) або відносна анемія з гемоглобіном менше 160 г/л; ретикулоцити і гематокрит підвищені. З віком розвивається поліцитемія, поліглобулія, що сприяють збільшенню кисневої ємності крові.
Ускладнення:
Тривало існуюча і виражена артеріальна гіпоксемія сприяє затримці розумового розвитку та органічному ураженню ЦНС.
Одним із ускладнень може бути абсцес мозку, який виникає шляхом переходу гнійного процесу з внутрішнього вуха, носових пазух в головний мозок. Можливі також посттравматичні і метастатичні абсцеси (при бактеріальному ендокардиті, після екстракції зуба, видалення мигдаликів), нагноєння старих вогнищ інфаркту або розмўякшення мозку. Ці ускладнення частіше зустрічаються в віці від 2-х до 20 років.
Одним із післяопераційних ускладнень після операції є бактеріальний ендокардит. Причиною його також можуть бути гнійні захворювання шкіри, каріозні зуби, хронічний тонзиліт та гайморит.
Лікування:
Хірургічне лікування рекомендується всім хворим з тетрадою Фалло.
Паліативні операції дозволяють хворим досягнути віку 5-6 років, коли з меншим ризиком можна виконати радикальну операцію.
Найбільш поширені типи паліативних втручань: системно-легеневі або міжартеріальні анастомози Блелока-Тауссінг (підключично-легеневий анастомоз), Ватерстоуна-Кулі (внутрішньоперикардіальний анастомоз між висхідною аортою і правою легеневою артерією), з’єднання даних судин за допомогою біологічних анастомозів (анастомоз Вишневського-Донецького) або синтетичних протезів, і в меншій мірі анастомоз Поттса-Сміта (анастомоз між низхідною аортою лівою легеневою артерією).
Радикальна корекція: закриття ДМШП і усунення стенозу легеневої артерії. Також необхідно усунути раніше виконані міжартеріальні анастомози. Радикальну корекцію тетради Фалло в ранньому віці слід проводити хворим, у яких стеноз вивідної протоки ПШ обумовлений головним чином зміщенням і гіпертрофією конусної перегородки при достатньо широкому легеневому стовбурі і артеріях. Радикальну операцію після виконаного в ранньому віці анастомозу по Блелоку-Тауссінгу доцільно виконувати через 1-3 роки, бажано до досягнення дитиною шкільного віку.
Прогноз:
Середня тривалість життя неоперованих хворих – близько 12 років. Однак близько 10% з них досягають 20 років, близько 1% – 30 років, у рідких випадках хворі досягають 75 років.
Летальність після паліативних операцій коливається від 1,6 до 15,6% в залежності від анастомозу у хворого і його віку.
Летальність після радикальних операцій коливається від 9,3 до 16,9%. Віддалені наслідки радикального лікування неускладнених форм тетради Фалло, як правило, хороші: через 10 років після операції 95% пацієнтів ведуть практично нормальне життя. 93-96% пацієнтів виписаних з клініки живуть більше 10 років, а 91% – більше 20 років.
Акушерська тактика:
При пренатальному виявлені тетради Фалло необхідно ретельно обстежити плід на предмет наявності у нього інших вад серця та внутрішніх органів та рекомендувати амніоцентез для хромосомного аналізу. При виявлені води до періоду життєздатності плода слід запропонувати переривання вагітності. У випадку пролонгування вагітності родорозрішення слід проводити в спеціалізованому центрі, в якому може бути надана невідкладна допомога лікарем педіатром-кардіологом.
Профілактика:
Попередження впливу шкідливих факторів, які можуть вплинути на ембріогенез (захворювання вагітної, неповноцінне харчування, гіпоксія, хімічні і радіологічні фактори і т.д.).
Номер з каталогу МІМ:
187501 Tetralogy of Fallot and Glaucoma
Літературні джерела:
- Белоконь Н.А., Подзолков В.П. Врожденные пороки серца.- М.: Медицина,1991,- C. 177-189.
- Кардиология детского возраста /Под ред. Мощича П.С., Сидельникова В.М., Кривчени Д.Ю.- К.: Здоровья,1986.- C. 159-165.
- Пренатальная диагностика вродженных пороков развития /Р.Ромеро, Д.Пипу, Ф.Дженти, Дж.С.Хоббинс.- М.: Медицина,1994.- C. 160-163.
- Шабалов Н.П. Неонаталогия /В 2-х томах.- СПб.: Специальная литература,1997.- Том 2.- С. 220-223.
Переглянуто редакційною колегією I.B.I.S.: 05/02/2002
Трисомія 18 – зріст
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”
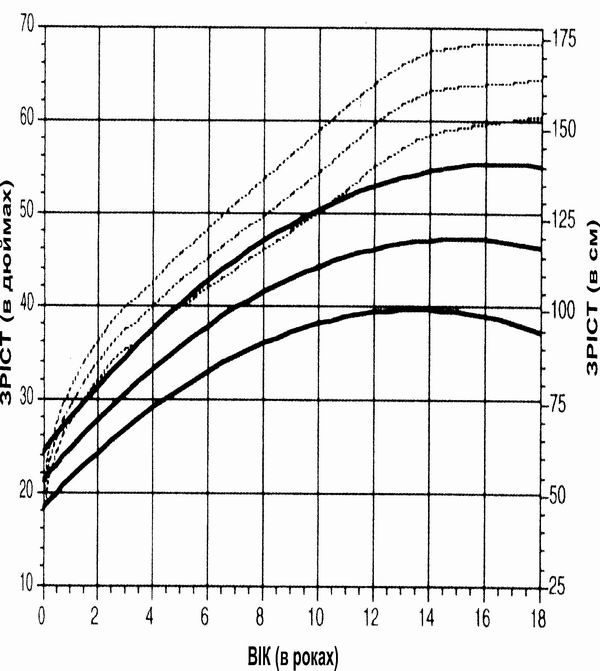
Криві збільшення зросту в пацієнтів з Трисомією 18 від народження до 18-річного віку (суцільні лінії) порівнюються з даними розвитку здорових осіб (пунктирні лінії). Вимірювання (84% – дівчатка) проводились у 76 пацієнтів. Baty BJ et al. Natural History of Trisomy 18 and Trisomy 13:I. Growth, Physical Assessment, Medical Histories, Survival, and Recurrence Risk. Am J Med Genet 49:175-188 (1994) Copyright (c) 1994 Wiley-Liss, Inc. Reprinted by permission of Wiley-Liss, Inc., a division of John Wiley & Sons, Inc.
Тератома шиї
(Neck Teratoma)
Олег Кравчук
Лікар акушер-гінеколог
Рівненського пологового будинку №2
Визначення:
Шийна тератома (тиреоїдна тератома) – пухлина з ембріональних клітин, яка локалізується в шийній ділянці.
Частота: невідома.
Співвідношення статей:
Cтатевих відмінностей в захворюванні не відмічено.
Етіологія:
Аномалія зустрічається спорадично, її причини невідомі.
Патологічна анатомія:
В 90% випадків тератоми діагностуються при народженні або після нього. Пухлини бувають частіше однобічними та інкапсульованими. Їхні розміри варіюють. Зазвичай вони складаються з кистозного та солідного компонентів. Малігнізуються вкрай рідко, а повідомлення про рецидив захворювання після радикального хірургічного лікування в сучасній літературі відсутні. Кальцинація виявлена в 50% випадків. Переважаючим гістологічним компонентом буває нервова тканина. Обструкція дихальних шляхів тканиною пухлини може призводити до гострої дихальної недостатності в неонатальному періоді. Багатовіддя, як ускладнення, зустрічається в 30% випадків і вважається результатом обструкції стравоходу. Виявлена безсумнівна кореляція між розмірами пухлини і багатовіддям.
Асоційовані вади:
Поєднані аномалії зазвичай не зустрічаються. У деяких випадках відмічали гіпоплазію легень, неперфорований анус, трисомію 13, хондродистрофію.
Диференційний діагноз:
Діагностика базується на виявленні утворів складної будови в шийній ділянці, які диференціюють від кистозної гігроми, зобу, шийного менінгоцеле, гемангіоми шийної ділянки. Кистозні гігроми – це кистозні утвори з типовою комірчастою будовою. Шийні менінгоцеле визначаються у вигляді утворів, при цьому визначаються спінальні дефекти. Шийні менінгоцеле можуть бути змішаної будови, однак їх локалізація і супутні спінальні дефекти можуть служити допоміжними діагностичними ознаками. Зоб має солідну будову, зазвичай не містить кистозного компоненту, розміщується симетрично в передніх відділах шиї і не досягає розмірів тератом. Бранхіогенні кисти представлені чисто кистозними утворами, розміщеними на передній поверхні грудинно-ключично-соскоподібного м’язу. Гемангіоми можуть бути представлені як кистозними, так і солідними утворами. Проведення диференційної діагностики з мезенхімальними пухлинами може виявитись неможливим, так як вони звичайно являють собою солідні пухлини. Нейробластоми шиї зазвичай відрізняються змішаною структурою. Багатовіддя буває рідко, кальцинація зустрічається в 40-45% випадків.
Прогноз:
За даними літератури, частота мертвонароджуваності при цій патології складає 17%. Смертність серед нелікованих новонароджених варіює від 80 до 100%. Основною причиною смерті є обструкція верхніх дихальних шляхів. Операційна смертність складає 9-15%. У більшості випадків пухлини бувають доброякісними, рецидивів після їх тотального видалення в періоді новонародженості не відмічалось.
Акушерська тактика:
Пухлини великого розміру можуть стати причиною клінічно вузького тазу. В цьому випадку показано абдомінальне родорозрішення. При пухлинах малого розміру притримуються стандартної акушерської тактики. Показане проведення динамічного ультразвукового дослідження для оцінки росту пухлини та зміни об’єму навколоплідних вод. До необхідних умов відносять родорозрішення в пренатальних центрах третього рівня. При цьому бригада лікарів повинна бути готова до інтубації дитини безпосередньо після її народження. При вираженому багатовідді може бути проведена амніографія в якості непрямого методу оцінки ступеня трахеоезофагальної обструкції.
Література:
- Пренатальная диагностика врожденных пороков развития плода /Р. Ромеро, Дж. Пилу, Ф. Дженти, А. Гидини, Дж.С. Хоббинс.- М.: Медицина,1994.- С. 124-126.
- Benacerraf BR. Ultrasound of Fetal Syndromes. Churchill Livingstone. 1998:351-356.
Переглянуто редакційною колегією I.B.I.S.: 27/08/2003
Трисомія 18 – вага
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”

Криві збільшення ваги в пацієнтів з Трисомією 18 від народження до 18-річного віку (суцільні лінії) порівнюються з даними розвитку здорових осіб (пунктирні лінії). Вимірювання (84% – дівчатка) проводились у 76 пацієнтів. Baty BJ et al. Natural History of Trisomy 18 and Trisomy 13:I. Growth, Physical Assessment, Medical Histories, Survival, and Recurrence Risk. Am J Med Genet 49:175-188 (1994) Copyright (c) 1994 Wiley-Liss, Inc. Reprinted by permission of Wiley-Liss, Inc., a division of John Wiley & Sons, Inc.
|
|
|
|
|
|




