
Serhiy
Синдром Прадера-Віллі – довжина середнього пальця руки
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”.
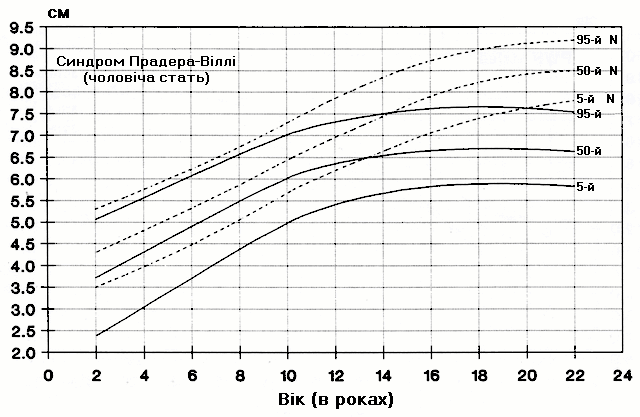
Стандартизовані криві довжини середнього пальця руки (95-й, 50-й та 5-й персентилі) у осіб з синдромом Прадера-Віллі (42 хлопчики і 29 дівчат) порівнюються з нормою (N). Було обстежено пацієнтів з видимою цитогенетичною делецією (52%), нормальним каріотипом (37%) та невідомим хромосомним статусом (11%). Приблизно половина обстежених пацієнтів була на низькокалорійній дієті. Деяки пацієнти спостерігались протягом 6 років. Butler MG and Meaney FJ: Pediatrics 88:853, 1991. Copyright © 1991 Reproduced by permission of Pediatrics.
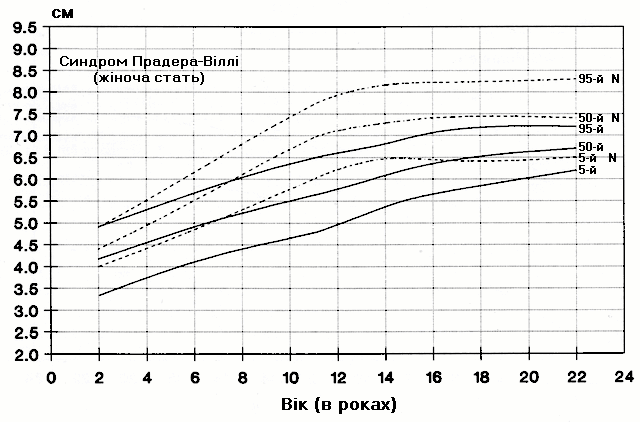
Синдром Прадера-Віллі – довжина кисті руки
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”.

Стандартизовані криві довжини кисті руки (95-й, 50-й та 5-й персентилі) у осіб з синдромом Прадера-Віллі (42 хлопчики і 29 дівчат) порівнюються з нормою (N). Було обстежено пацієнтів з видимою цитогенетичною делецією (52%), нормальним каріотипом (37%) та невідомим хромосомним статусом (11%). Приблизно половина обстежених пацієнтів була на низькокалорійній дієті. Деяки пацієнти спостерігались протягом 6 років. Butler MG and Meaney FJ: Pediatrics 88:853, 1991. Copyright © 1991 Reproduced by permission of Pediatrics.
Синдром Прадера-Віллі – обвід голови у дівчат
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”.

Стандартизовані криві збільшення обводу голови (95-й, 50-й та 5-й персентилі) у 29 осіб з синдромом Прадера-Віллі порівнюються з нормою (N). Було обстежено 71 пацієнта з подібним клінічним діагнозом (42 хлопчики та 29 дівчат, включаючи тих, в кого спостерігались цитогенетична делеція (52%), нормальний каріотип (37%) та невідомий хромосомний статус (11%). Приблизно половина обстежених пацієнтів була на низькокалорійній дієті. Деяки пацієнти спостерігались протягом 6 років. Butler MG and Meaney FJ: Pediatrics 88:853, 1991. Copyright © 1991 Reproduced by permission of Pediatrics.
Синдром Прадера-Віллі – зріст дівчат
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”.

Стандартизовані криві збільшення зросту (95-й, 50-й та 5-й персентилі) у 29 осіб з синдромом Прадера-Віллі порівнюються з нормою (N). Було обстежено 71 пацієнта з подібним клінічним діагнозом (42 хлопчики та 29 дівчат, включаючи тих, в кого спостерігались цитогенетична делеція (52%), нормальний каріотип (37%) та невідомий хромосомний статус (11%). Приблизно половина обстежених пацієнтів була на низькокалорійній дієті. Деяки пацієнти спостерігались протягом 6 років. Butler MG and Meaney FJ: Pediatrics 88:853, 1991. Copyright © 1991 Reproduced by permission of Pediatrics.
Синдром Прадера-Віллі – вага дівчат
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”.
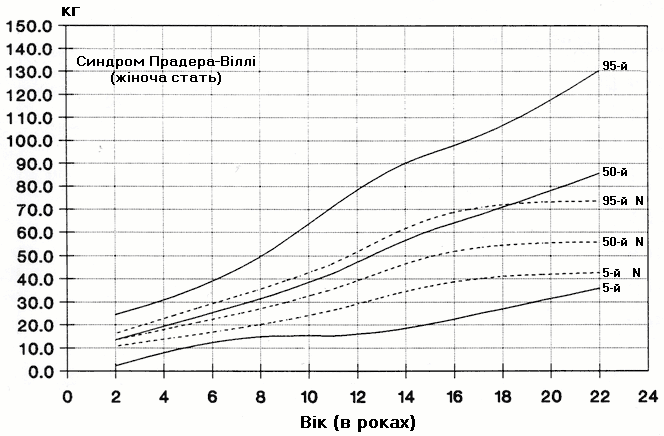
Стандартизовані криві збільшення ваги (95-й, 50-й та 5-й персентилі) у 29 осіб з синдромом Прадера-Віллі порівнюються з нормою (N). Було обстежено 71 пацієнта з подібним клінічним діагнозом (42 хлопчики та 29 дівчат, включаючи тих, в кого спостерігались цитогенетична делеція (52%), нормальний каріотип (37%) та невідомий хромосомний статус (11%). Приблизно половина обстежених пацієнтів була на низькокалорійній дієті. Деяки пацієнти спостерігались протягом 6 років. Butler MG and Meaney FJ: Pediatrics 88:853, 1991. Copyright © 1991 Reproduced by permission of Pediatrics.
Синдром Прадера-Віллі – обвід голови у хлопчиків
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”.
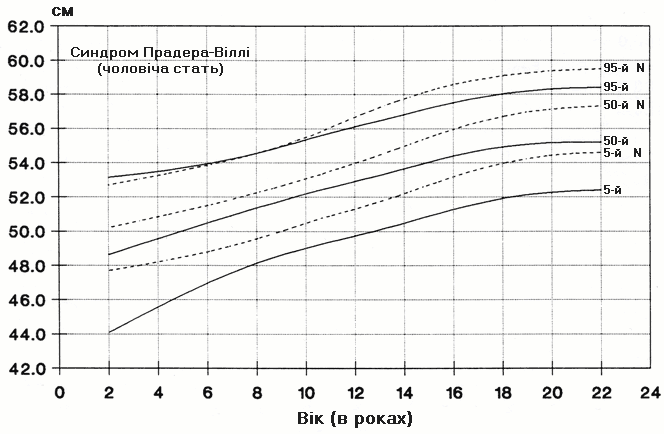
Стандартизовані криві збільшення обводу голови (95-й, 50-й та 5-й персентилі) у 42 осіб європеоїдної раси з синдромом Прадера-Віллі порівнюються з нормою (N). Було обстежено 71 пацієнта з подібним клінічним діагнозом (42 хлопчики та 29 дівчат, включаючи тих, в кого спостерігались цитогенетична делеція (52%), нормальний каріотип (37%) та невідомий хромосомний статус (11%). Приблизно половина обстежених пацієнтів була на низькокалорійній дієті. Деяки пацієнти спостерігались протягом 6 років. Butler MG and Meaney FJ: Pediatrics 88:853, 1991. Copyright (c) 1991 Reproduced by permission of Pediatrics.
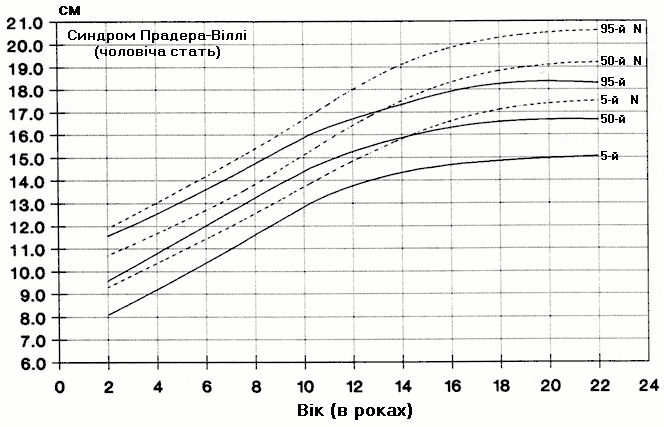
Синдром Прадера-Віллі – зріст хлопчиків
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”.
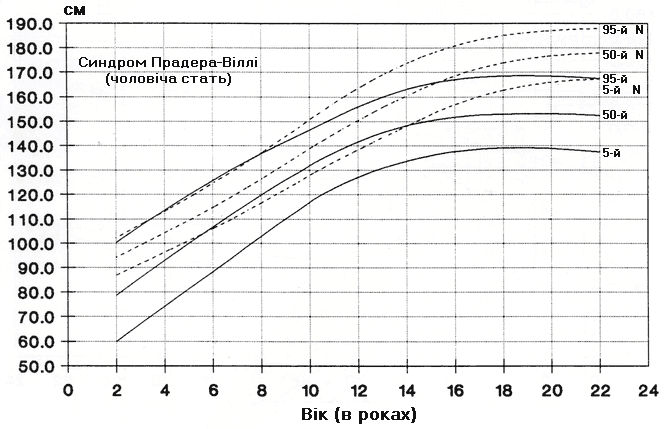
Стандартизовані криві збільшення зросту (95-й, 50-й та 5-й персентилі) у 42 осіб європеоїдної раси з синдромом Прадера-Віллі порівнюються з нормою (N). Було обстежено 71 пацієнта з подібним клінічним діагнозом (42 хлопчики та 29 дівчат, включаючи тих, в кого спостерігались цитогенетична делеція (52%), нормальний каріотип (37%) та невідомий хромосомний статус (11%). Приблизно половина обстежених пацієнтів була на низькокалорійній дієті. Деяки пацієнти спостерігались протягом 6 років. Butler MG and Meaney FJ: Pediatrics 88:853, 1991. Copyright (c) 1991 Reproduced by permission of Pediatrics.
Синдром Прадера-Віллі – вага хлопчиків
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”.

Стандартизовані криві збільшення ваги (95-й, 50-й та 5-й персентилі) у 42 осіб європеоїдної раси з синдромом Прадера-Віллі порівнюються з нормою (N). Було обстежено 71 пацієнта з подібним клінічним діагнозом (42 хлопчики та 29 дівчат, включаючи тих, в кого спостерігались цитогенетична делеція (52%), нормальний каріотип (37%) та невідомий хромосомний статус (11%). Приблизно половина обстежених пацієнтів була на низькокалорійній дієті. Деяки пацієнти спостерігались протягом 6 років. Butler MG and Meaney FJ: Pediatrics 88:853, 1991. Copyright (c) 1991 Reproduced by permission of Pediatrics.
Синдром Прадера-Віллі
(Prader-Willi Syndrome)
Т.Г. Гаркун
Інформаційний спеціаліст Хмельницького ОМНІ-центру
Що таке синдром Прадера-Віллі?
Це складне генетичне захворювання, що характеризується низькою статурою, затримкою розумового розвитку або труднощами у навчанні, неповноцінним статевим розвитком. Пацієнти мають певні проблеми у поведінці. У них низький тонус м’язів. Хворі на синдром Прадера-Віллі мають нестримне бажання постійно їсти, що, поряд зі зниженою потребою в калоріях, призводить до ожиріння.
Чи успадковується даний синдром?
Виникнення синдрому Прадера-Віллі пов’язане із мікроаномалією 15 хромосоми, що, переважно, трапляється спонтанно.
Як часто зустрічається така проблема?
У США 1 з 14000 людей має синдром Прадера-Віллі, а у новонароджених це захворювання зустрічається ще частіше. Синдром Прадера-Віллі є одним із 10 найрозповсюдженіших генетичних захворювань та основною генетичною причиною ожиріння.
Проблеми з їжею, що пов’язані з синдромом Прадера-Віллі, починаються з народження?
Ні. Новонароджених з даним синдромом часто описують, як “млявих” діток, які не можуть активно смоктати материнське молоко, щоб отримати всі необхідні поживні речовини через знижений тонус м’язів (гіпотонію). Дуже часто їх годують через зонд протягом перших кількох місяців після народження, поки вони не зможуть контролювати м’язи. Пізніше, ближче до дошкільного віку, у дітей із синдромом Прадера-Віллі з’являється підвищений інтерес до їжі, і вони швидко набирають зайву вагу, якщо не обмежувати калорії.
Чому хворі на синдром Прадера-Віллі їдять так багато?
Хворі на даний синдром мають дефект у частині мозку (гіпоталамусі), що відповідає за голод та відчуття ситості. Такі люди ніколи не відчувають себе цілком ситими, тому мають постійне бажання їсти. До того ж, хворі з синдромом Прадера-Віллі потребують менше калорій, ніж здорові для підтримки нормальної ваги. Ожиріння є основною причиною важкого перебігу хвороби та смерті при синдромі Прадера-Віллі. Як і для всіх людей, ожиріння при даному синдромі може призвести до підвищення артеріального тиску, проблем з диханням, діабету, тощо.
Чи можна щось зробити, щоб контролювати постійне бажання їсти?
На жаль, таблетки, що знижують апетит, допомагають хворим на синдром Прадера-Віллі лише тимчасово. Більшість хворих усе життя повинні дотримуватись низькокалорійної дієти і мати обмежений доступ до їжі. Наприклад, багато сімей змушені закривати кухні та холодильники на замок.
Я знаю багато людей, які їдять багато та мають ожиріння. Як дізнатись, чи у них немає синдрому Прадера-Віллі?
Із даним синдромом пов’язане не тільки ожиріння. Хворі мають характерну зовнішність, проблеми з мовленням, труднощі у навчанні, затримку розумового розвитку, і ще ряд проблем. Ці ознаки повинні бути присутніми, коли клінічно діагностується синдром Прадера-Віллі. Спеціальні генетичні тести можуть підтвердити цей діагноз.
Які генетичні причини виникнення цього синдрому?
Синдром Прадера-Віллі виникає через відсутність декількох генів на одній з хромосом, які зокрема, відповідають за правильне функціонування гіпоталамусу. Цю проблему активно досліджують у лабораторіях всього світу, тому що розуміння даного синдрому не тільки допоможе людям із синдромом Прадера-Віллі, але й дозволить глибше зрозуміти механізми виникнення ожиріння, затримки розумового розвитку або проблем з поведінкою у інших.
А які проблеми з поведінкою є у людей із даним захворюванням?
Крім того, що хворі на цей синдром іноді використовують будь-які екстремальні спроби дістати їжу, вони часто є роздратованими, впертими, жорстокими, люблять сперечатися, мають нав’язливі думки та вчинки. З такими людьми потрібно поводитись обережно та використовувати спеціальні технології для корекції характеру та поведінки, іноді потрібна медикаментозна терапія.
Як впливає синдром Прадера-Віллі на мовлення хворих?
Синдром Прадера-Віллі ускладнює всі аспекти розвитку мови та мовлення. Коли інтелектуальні здібності є обмеженими, рецептивне і експресивне мовлення порушується, і здатність правильно вимовляти окремі звуки (артикуляція) також страждає.
Такі проблеми з мовленням у хворих на даний синдром можуть виникати у зв’язку з нейромоторними проблемами, такими як гіпотонія (низький тонус м’язів). Гіпотонія впливає на швидкість мовлення, якість звуків, здатність координувати рухами язика, губів, щелеп та піднебіння. Тому мовлення у хворих на синдром Прадера-Віллі може бути сповільненим, невиразним і/або гугнявим. У таких випадках пацієнтам слід займатись із логопедами за спеціальними методиками для покращання якості мовлення.
Чи можуть хворі на синдром Прадера-Віллі вести нормальне життя?
Вони можуть відвідувати школу, охоче приймати участь у колективній діяльності, отримувати роботу, навіть далеко від дому. Однак, вони постійно потребують сторонньої допомоги. У шкільному віці вони потребують спеціальних методик навчання та програм для покращання мовлення, постійного контролювання у прийомах їжі. Хоча більшість хворих на синдром Прадера-Віллі помирають у підлітковому або молодому віці, теоретично вважається, що, якщо не допустити розвитку ожиріння, то можна вести нормальне життя.
Переглянуто редакційною колегією I.B.I.S.: 25/06/2005
Дивіться також:
- Синдром Прадера-Віллі (інформація для спеціалістів)
- Синдром Прадера-Віллі (діаграми розвитку дітей):
Синдром Прадера-Віллі
(Prader-Willi Syndrome)
О.Г. Дуліпа
Лікар-генетик
Старокостянтинівської центральної районної лікарні Хмельницької області
Вперше синдром був описаний швейцарськими педіатрами A. Prader та H. Willi у 1956 році.
Основні діагностичні критерії:
- гіпотонія;
- гіпогонадизм;
- ожиріння;
- розумова відсталість.
Клінічна картина:
Новонароджені, зазвичай, народжуються з вираженою м’язовою гіпотонією, часто відмічається зниження рухової діяльності в утробі матері.
Сухожильні рефлекси, рефлекс Моро, ковтальний та смоктальний рефлекси знижені, що приводить до труднощів при годуванні та респіраторних ускладнень. Такі пацієнти малорухливі. Протягом 2-го – 3-го року життя гіпотонія зменшується, а до шкільного віку майже зникає.
Ожиріння у пацієнтів розвивається через декілька тижнів або місяців внаслідок вираженої поліфагії. Характерне постійне відчуття голоду. Поглинання їжі часто і в великій кількості є домінуючим проявом синдрому. Надлишок жиру відкладається в нижній частині тулуба та сідницях, в проксимальній частині кінцівок. Висловлюється припущення, що ожиріння у хворих зумовлено значним (більше ніж у 10 разів) посиленням синтезу жиру із ацетату і надзвичайно низькими процесами ліполізу.
У чоловіків спостерігається гіпогонадизм: гіпоплазія статевого члена, калитки, крипторхізм. Біопсія сім’яників показала відсутність гермінальних клітин при наявності клітин Сертолі.
У жінок спостерігається аменорея, в переважній більшості випадків гіпоплазія матки. Рівень гонадотропінів та статевих стероїдів зменшений в усіх випадках. Гіпогонадизм по гіпогонадотропному типу може бути пов’язаний з дисфункцією гіпоталамуса переважно в районі вентромедіального та вентролатерального ядер. Правильність цієї точки зору підтверджується ефективністю лікування хворих фармацевтичними препаратами (кломіфен), що приводить до збільшення в плазмі вмісту лютеїнізуючого гормону, тестостерону, нормалізації показників ниркової екскреції гонадотропінів, сперматогенезу та виникненню вторинних статевих ознак.
Крім основних ознак зустрічається мікроцефалія, зменшений біфронтальний діаметр, косоокість, мигдалеподібні очі, сколіоз. Стопи і кисті диспропорційно малі (акромікрія). Пацієнти низького росту, мають гіпопігментацію шкіри, волосся. Одним із пояснень гіпопігментації є зниження активності тирозинази у волосяних фолікулах та меланоцитах, а також зменшення пігменту в сітківці. У таких хворих відмічається діабетичний тип толерантності до глюкози.
Більшість пацієнтів страждає на розумову відсталість. Рівень розумового розвитку в межах від 20 до 90 IQ. Відмічається затримка мовного розвитку, словниковий запас обмежений; характерна емоційна неврівноваженість.
Зустрічаються й інші аномалії: мікродонтія, гіпоплазія хрящів вушних раковин, сколіоз, ектропіон, глаукома.
При морфологічному дослідженні мозку та ядерно-магнітному резонансі (ЯМР) можуть спостерігатися приблизно у 12% кісти хробака мозочка та аномалії кори головного мозку.
Тип успадкування:
Автори, які вперше описали синдром, висунули припущення про аутосомно-рецесивний тип успадкування захворювання. Згодом з’явились повідомлення про можливість аутосомно-домінантного типу успадкування. Підтвердженням таких гіпотез могли бути сімейні випадки патології. Однак більшість описаних клінічних спостережень синдрому Прадера-Віллі мали спорадичний характер. Наступні дослідження дозволили встановити у дітей із синдромом Прадера-Віллі певні хромосомні порушення. Цитогенетичний аналіз показав, що хромосомні аномалії у хворих були представлені або транслокаціями (t 15/15), або мозаїцизмом. У 1987 році з’явилися перші повідомлення про мікроделеції хромосоми 15. Однак кінцева ідентифікація хромосомних змін при синдромі Прадера-Віллі стала можливою лише після введення в практику молекулярно-генетичних методів дослідження.
Патогенез:
Розвиток цього захворювання пояснюється такими генетичними явищами, як геномний імпринтинг та однобатьківська (уніпарентна) дисомія.
Геномний імпринтинг означає різну експресію генетичного матеріалу (гомологічних алелей) в хромосомах залежно від їх батьківського чи материнського походження, тобто свідчить про вплив батьків на фенотип дитини. До теперішнього часу вважалось, що експресія генів батька та матері рівноцінна. Геномний імпринтинг – це статевий та залежний від тканини складний модифікатор генної активності деяких локусів хромосом залежно від їх батьківського походження. Феномен геномного імпринтингу виявлений і при інших захворюваннях – синдромі Сотоса, Беквіта-Відемана, Сільвера-Рассела, муковісцидозі та інших.
Уніпарентна (однобатьківська) дисомія – успадкування обох хромосом лише одного із батьків. Протягом багатьох років вважалось, що таке успадкування неможливе. Лише з допомогою молекулярно-генетичних маркерів вдалося довести можливість однобатьківської дисомії. Природа уніпарентної дисомії остаточно не встановлена, однак відомо, що її причиною є низка генетичних та біохімічних порушень.
Хромосомні механізми, що відповідають за синдром Прадера-Віллі можуть бути різного походження. Один з них викликаний делецією батьківського критичного сегмента 15q 11.2-q12, або ж через втрату цілої батьківської 15 хромосоми (уніпарентна материнська дисомія). Подібні зміни спостерігаються у 75% випадків. І навпаки – відсутність материнської частини хромосоми (уніпарентна батьківська дисомія) викликає інший характерний фенотип – синдром Ангельмана (ангелоподібний), який зустрічається значно рідше – 25%. Більшість хворих мають нормальний каріотип, але у деяких спостерігається або транслокація 15/15, або транслокація між хромосомою 15 та іншою аутосомою. За допомогою високоточного хромосомного аналізу та FISH-методу, можна виявити мікроскопічну делецію батьківської частини хромосоми (SNRPh – 182279).
На основі патогенезу Олаєцер та співавтори (2000 р.) вказали на випадки трьох фенотипів синдрому Прадера-Віллі:
- Пацієнти з вираженою батьківською хромосомою мають типовий фенотип синдрому Прадера-Віллі.
- Пацієнти з материнською уніпарентною дисомією мають більш легкий фенотип з кращою пізнавальною функцією.
- Пацієнти з материнською уніпарентною дисомією і мозаїчною 15 трисомією мають більш виражений фенотип з великою кількістю вроджених серцевих захворювань.
Ризик для сибсів пробанда:
Якщо уражена одна дитина і каріотип абсолютно нормальний, то ступінь ризику для потомства 0,4%, якщо ж 2 і більше сибсів уражені, то ризик відповідно становить 50%.
Прогноз:
Прогноз відносно життя сприятливий і визначається важкістю протікання цукрового діабету та кардіореспіраторними ускладненнями через важку форму ожиріння.
Слід звернути увагу на підвищений ризик розвитку лейкемії у хворих з синдромом Прадера-Віллі. Дослідження показали, що відбувається зниження репарації ДНК (до 65% порівняно з 97% у здорової дитини) в лімфоцитах хворих з цією патологією. Не виключено, що низька репараційна спроможність ДНК може спричинити розвиток злоякісних новоутворень у пацієнтів із синдромом Прадера-Віллі.
Диференційний діагноз:
Диференційний діагноз проводиться із синдромом Лоуренса-Муна-Барде-Бідля, адіпозогенітальною дистрофією, міопатією вродженою, аміотрофією спінальною вродженою, Альстрема синдромом.
Популяційна частота:
За даними реєстру асоціації хворих із синдромом Прадера-Віллі в США та Канаді нараховується понад 2000 хворих. В останні роки вдалося встановити популяційну частоту патології, що становить 1:10000 – 1:20000 серед живонароджених.
Вік маніфестації: з народження.
Співвідношення статі: Ч 1 : Ж 1.
Лікування:
- Контрольований режим харчування з врахуванням калорійності їжі.
- Лікування цукрового діабету оральними гіпоглікемічними препаратами.
- Надання допомоги при кардіореспіраторних ускладненнях.
- Відновлення секреції гонадотропінів і статевих стероїдів за допомогою кломіфену.
Профілактика:
Медико-генетичне консультування родин, де є пробанд із синдромом Прадера-Віллі з проведенням комплексу молекулярно-цитогенетичних досліджень.
Номер з каталогу МІМ:
176270 Prader-Willi Syndrome; PWS
601491 Imprinted in Prader-Willy Syndrome; IPW
264010 Prader-Willi Habitus, Osteopenia, and Camptodactyly
600161 Prader-Willi/Angelman Region RNA1; PWAR1
Література:
- Наследственные синдромы и медико-генетическое консультирование / Е.И. Козлова, О. Семенова, Н.С. Демидова, О.Е. Блинникова.- Ленинград: Медицина, 1997.
- Butler JV, Whittington JE, Holland AJ, Boer H, Clarke D, Webb T. Prevalence of, and risk factors for, physical ill-health in people with Prader-Willi syndrome. Dev Med Child Neurol 2002; Apr 44(4):248-55.
- Goldberg DL, Garrett CL, Van Riper C, Warzak WJ. Coping with Prader-Willi syndrome. J Am Diet Assoc 2002; Apr 102(4):537-42.
- Ledbetter DH, Riccardi VM, Airhart SD, Strobel RJ, Keenan BS, Crawford JD. Deletions of chromosome 15 as a cause of the Prader-Willi syndrome. New Eng. J. Med 1981;304:325-329.
- Tobias ES, Tolmie JL, Stephenson JB. Cataplexy in the Prader-Willi syndrome. Arch Dis Child 2002;Aug 87(2):170.
- Zellweger H, Schneider HJ. Syndrome of hypotonia-hypomentia-hypogonadism-obesity (HHHO) or Prader-Willi syndrome. Am. J. Dis. Child 1968;115:588-598.
Переглянуто редакційною колегією I.B.I.S.: 08/07/2002
Дивіться також:
- Синдром Прадера-Віллі (інформація для батьків)
- Синдром Прадера-Віллі (діаграми розвитку дітей):
|
|
|
|
|
|






{kind=link}