
Uncategorized
Довжина тулуба
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”
3, 50 та 97 перцентилі довжини тулуба відповідно до гестаційного віку. Довжина тулуба визначаються відстанню між рукояткою грудини та найвищою точкою лобкового зчленування. Вимірювання проводились у 87 доношених (48 хлопчиків та 39 дівчаток) та 111 недоношених (55 хлопчиків та 56 дівчаток) ізраїльських новонароджених 27-41 тижнів гестації у перші 36-60 годин життя. Жодна дитина не мала вроджених вад. Merlob P et al: Birth Defects: Orig Art Series XX(7):30, 1984.
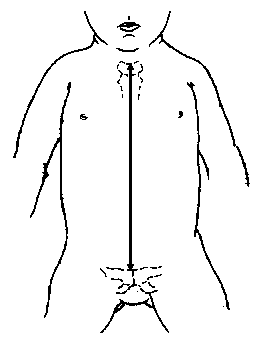

3, 50 та 97 перцентилі індексу довжини тулуба відповідно до гестаційного віку.
Індекс довжини тулуба =
Загальна довжина тулуба
Довжина тіла
Див. пояснення вище. Merlob P et al: Birth Defects: Orig Art Series XX(7):30, 1984.

Відстань між сосками
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”

3, 50 та 97 перцентилі відстані між сосками відповідно до гестаційного віку. Міжсоскові вимірювання визначаються відстанню між центрами обох сосків. Вимірювання проводились у 87 доношених (48 хлопчиків та 39 дівчаток) та 111 недоношених (55 хлопчиків та 56 дівчаток) ізраїльських новонароджених 27-41 тижнів гестації у перші 36-60 годин життя. Жодна дитина не мала вроджених вад. Merlob P et al: Birth Defects: Orig Art Series XX(7):27, 1984.

3, 60 та 97 перцентилі індексу відстані між сосками нанесені на графік відповідно до гестаційного віку.
Індекс відстані між сосками =
Відстань між сосками * 100
Окружність грудей
Див. пояснення вище. Merlob P et al: Birth Defects: Orig Art Series XX(7):27, 1984.
Виміри тулуба
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”
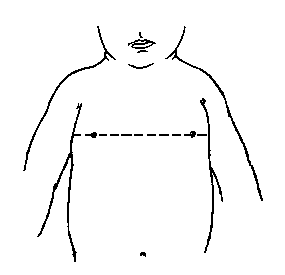 3, 50 та 97 перцентилі обводу грудей відповідно до гестаційного віку. Обвід грудей вимірюється стрічкою, яку розміщують на рівні сосків, коли дитина знаходиться в спокійному стані. Вимірювання проводились у 87 доношених (48 хлопчиків та 39 дівчаток) та 111 недоношених (55 хлопчиків та 56 дівчаток) ізраїльських новонароджених 27-41 тижнів гестації у перші 36-60 годин життя. Жодна дитина не мала вроджених вад. Merlob P et al: Birth Defects: Orig Art Series XX(7):26, 1984.
3, 50 та 97 перцентилі обводу грудей відповідно до гестаційного віку. Обвід грудей вимірюється стрічкою, яку розміщують на рівні сосків, коли дитина знаходиться в спокійному стані. Вимірювання проводились у 87 доношених (48 хлопчиків та 39 дівчаток) та 111 недоношених (55 хлопчиків та 56 дівчаток) ізраїльських новонароджених 27-41 тижнів гестації у перші 36-60 годин життя. Жодна дитина не мала вроджених вад. Merlob P et al: Birth Defects: Orig Art Series XX(7):26, 1984.

Міжсосковий індекс
![]()
Міжсосковий індекс визначався у 506 морфологічно здорових новонароджених 26-44 тижнів гестації в Угорщині. Виключались діти із великими вродженими аномаліями і непевним гестаційним віком. Вимірювання проводились у перші 5 днів життя. Міжсосковий індекс виявився незмінним для всіх значень гестаційного віку (25,1 ± 2,9). Якщо міжсосковий індекс був більший, ніж 28,0, вважалось, що соски розміщені широко. Mehes K et al: J Pediatr 85:91, 1974.
Розмір краю печінки
Відстань від краю печінки нижче реберного краю вимірювалась у 367 здорових доношених дітей. Вимірювання проводились у перші 26-44 години життя, потім повторно на 72-96 годинах життя. Вимірювання проводились від правого реберного краю на рівні міжключичної лінії. Гестаційний вік оглянутих дітей 36-41 тижнів.
Не виявлено жодних вагомих відмінностей у різного гестаційного віку чи залежно від часу дослідження. Середня відстань становила 3,0 см із стандартним відхиленням ±0,7 см. Дані є достатніми для встановлення гепатомегалії, коли печінка пальпується нижче, ніж 4,4 см від реберного краю. Ashkenazi S et al: Am J Dis Child 138:377, 1984.
Довжина фільтру
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”

3, 50 та 97 перцентилі довжини фільтру відповідно до гестаційного віку. Вимірювання проводились у 87 доношених (48 хлопчиків та 39 дівчаток) та 111 недоношених (55 хлопчиків та 56 дівчаток) ізраїльських новонароджених 27-41 тижнів гестації у перші 36-60 годин життя. Жодна дитина не мала вроджених вад. Merlob P et al: Birth Defects: Orig Art Series XX(7):5, 1984.
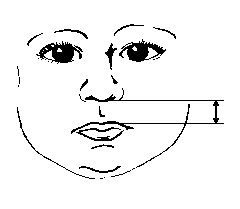

3, 50 та 97 перцентилі довжини фільтру відповідно до ваги тіла при народженні. Вимірювання проводились у 87 доношених (48 хлопчиків та 39 дівчаток) та 111 недоношених (55 хлопчиків та 56 дівчаток) ізраїльських новонароджених 27-41 тижнів гестації у перші 36-60 годин життя. Жодна дитина не мала вроджених вад. Merlob P et al: Birth Defects: Orig Art Series XX(7):5, 1984. Sivan Y et al: J Med Genet 22:414, 1985.
Ширина рота
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”
3, 50 та 97 перцентилі ширини рота відповідно до гестаційного віку. Ширина ротової щілини (відстань між комісурами) – відстань між двома хейліонами (точки кутів ротової щілини), коли дитина знаходиться в спокійному стані. Вимірювання проводились у 87 доношених (48 хлопчиків та 39 дівчаток) та 111 недоношених (55 хлопчиків та 56 дівчаток) ізраїльських новонароджених 27-41 тижнів гестації у перші 36-60 годин життя. Жодна дитина не мала вроджених вад. Merlob P et al: Birth Defects: Orig Art Series XX(7):5, 1984. Вперше графіки були надруковані Sivan Y et al: J Med Genet 20:130, 1983.


3, 50 та 97 перцентилі ширини рота відповідно до ваги при народженні. Вимірювання проводились у 87 доношених (48 хлопчиків та 39 дівчаток) та 111 недоношених (55 хлопчиків та 56 дівчаток) ізраїльських новонароджених 27-41 тижнів гестації у перші 36-60 годин життя. Жодна дитина не мала вроджених вад. Merlob P et al: Birth Defects: Orig Art Series XX(7):5, 1984. Sivan Y et al: J Med Genet 22:414, 1985.

Задні уретральні клапани
(Posterior Urethral Valves)
З.О. Сосинюк
лікар пренатальної УЗД медико-генетичної консультації
Рівненського обласного клінічного лікувально-діагностичного центру ім. В. Поліщука
Включення:
Задні уретральні клапани, низька обструктивна уропатія.
Визначення:
Низька обструкція (уретри) обумовлена наявністю мембраноподібних структур в задніх відділах уретри і характеризується дилятованою уретрою проксимальніше від місця обструкції, вираженим збільшенням сечового міхура та двобічним розширенням сечоводів і піелоектазією.
Частота:
Зустрічається виключно у плодів чоловічої статі.
Етіологія:
Аномалія розвитку уретри зустрічається спорадично. Хоча клапани уретри були виявлені при синдромі “сливового живота”, хромосомних дефектах, у близнюків, у рідних братів і сестер, що свідчить про генетичний характер захворювання.
Ембріогенез і патологічна анатомія:
Задня стінка уретри в нормі має дві уретровагінальні складки, які утворюються внаслідок міграції вольфової протоки між 4 та 6 тижнями гестації, і тягнуться вздовж від мюллерового горбка до зачатків купферовських чи бартолонієвих залоз. Уретральні клапани – гетерогенні ембріональні зародки.
За формою клапан може представляти собою плоску пластинку з отвором в центрі, напівкруглу пластинку з центрально або ексцентрично розташованими отворами, які мають форму балона, що роздувається під час сечовипускання.
Вони можуть бути одиночними і множинними.
Розрізняють 3 анатомічні типи уретральних клапанів за Янгом. Тип І – найчастіший варіант, клапани розміщені дистальніше сім’яного горбку, прикріплюються до бокової стінки уретри. Тип ІІ зустрічається рідко, являє собою складки на сім’яному горбку, які проксимально пересікають шийку сечового міхура, де діляться на пальцеподібні мембрани. Тип ІІІ – мембранозна діафрагма, яка закриває отвір уретри і локалізується дистальніше від сім’яного горбка. Останній тип зустрічається нечасто, характерний для летальних наслідків.
Клапани можуть бути тонкими, покритими лише перехідним епітелієм, або ж містять сполучну тканину, яка надає їм потовщеного вигляду. Внаслідок затруднення відтоку сечі при уретральних клапанах розвивається гіпертрофія трабекул і стінки сечового міхура.
Нерідко вони поєднуються з дисплазією нирок, з’являються патологічні рефлюкси (в 30-50% випадків розвивається везікоуретральний рефлюкс), затримка сечовипускання.
Асоціативні аномалії:
Задні уретральні клапани поєднуються з іншими аномаліями сечовивідних шляхів: мегацистік, мегауретер, гідронефроз, дивертикули сечоводів і розширення проксимального відділу уретри, подвоєння уретри, крипторхізм, гіпоспадія.
Аномалії розвитку інших органів включають в себе гіпоплазію трахеї, відкриту артеріальну протоку, загальні вади легеневої венозної системи, стеноз мітрального клапану, сколіоз, аномалії скелету і нижніх кінцівок, атрезію ануса. Зустрічаються у плодів з хромосомними аномаліями, в тому числі трисомії 18 і 13, делеції 2q і 69 XXY.
Пренатальна ультразвукова діагностика:
При ультразвуковому скануванні візуалізація уретральних клапанів неможлива, враховуючи їх малі розміри. Діагноз задніх уретральних клапанів може бути запідозреним при виявленні непрямих ультразвукових критеріїв обструкції сечовивідних шляхів на низькому рівні, а саме: мегацистік, мегауретер, гідронефроз і маловіддя у плодів чоловічої статі. Можлива візуалізація розширення проксимального відділу уретри, гіпертрофії стінки сечового міхура. Сечоводи розширені та звивисті і в деяких випадках визначається місце їх впадіння в сечовий міхур.
Ступінь піелоектазії може бути різною. Іноді при вираженій обструкції та нирковій дисплазії не визначається розширення ниркової миски, що обумовлено декомпресією ниркової лоханки та мисково – сечоводною атрезією при дисплазії нирок.
Діагностичними ультразвуковими критеріями дисплазії нирок є наявність множинних кіст і гіперехогенність паренхіми нирок як при зменшених, так і збільшених розмірах нирок.
Внаслідок витоку сечі в черевну порожнину або ж в периренальний простір іноді виникає сечовий асцит. Причиною є розрив сечового міхура, але в більшості випадків можлива транссудація сечі. Виражений сечовий асцит може призвести до атрофії м’язів передньої черевної стінки (синдром prune-belly).
Наростаюче маловіддя – несприятлива прогностична ознака. Маловіддя визначається не завжди і залежить від ступеня та тривалості обструкції.
Диференційний діагноз:
Проводиться з іншими обструктивними уропатіями: обструкція мисково-сечовивідного, везикуло-сечовивідного сполучень, первинний мегауретер, виражений везікуло-уретральний рефлюкс, а також з синдромом мегалоцистік – мікроколон – кишкової гіпоперистальтики.
Прогноз:
Час виникнення обструкції сечовивідних шляхів є вирішальним фактором у визначенні перебігу захворювання нирок. При пренатальній діагностиці патології прогноз досить серйозний. Такі новонароджені відносяться до групи високого ризику (32-50%) смерті від таких ускладнень: пневмомедіастінуму, пневмотораксу, пов’язаних з гіпоплазією легень, поєднаними аномаліями, нирковою недостатністю і ускладненнями від проведеної хірургічної декомпресії. У цих дітей часто виникає затримка фізичного розвитку. Внаслідок оперативного лікування можлива нормалізація функції нирок. Але хронічне ураження паренхіми може тривожити незалежно від проведеного лікування. Термінальна стадія наступає до віку 9-10 років від гіпоплазії, дисплазії нирок і хронічного пієлонефриту, які сприяють прогресуванню ниркової недостатності.
Акушерська тактика:
Ведення вагітності при низькій обструкції сечовивідних шляхів залежить від виявлення інших поєднаних аномалій, терміну вагітності, стану функції нирок.
Пошук поєднаних аномалій при ультразвуковому скануванні ускладнений через маловіддя. Можливість проведення амніоцентезу та кордоцентезу з тих самих причин ускладнені. При виявленні хромосомних або анатомічних дефектів, несумісних з життям, оптимальним методом вибору є переривання вагітності.
Важливим для оцінки стану плода є об’єм навколоплідних вод. При нормальній кількості останніх прогноз для плода, як правило, сприятливий. Якщо прогноз для плода несприятливий у зв’язку з вираженим маловіддям, дисплазією нирок та порушенням їх функцій, до періоду життєздатності плоду показана елімінація вагітності. Якщо прогноз сприятливий, то вирішення питань акушерської тактики залежить від терміну вагітності. При доношеній вагітності родорозрішення проводиться в регіональному перинатальному центрі, де проводиться огляд новонародженого урологом в перші години життя. Спосіб ведення пологів не залежить від пренатально встановленого діагнозу.
При встановленні діагнозу в період життєздатності плода з незрілими легенями необхідно проводити його щотижневе ультразвукове сканування. В таких випадках показана внутрішньоутробна профілактична декомпресія або ж вичікувальна тактика і родорозрішення при наростаючому маловідді.
Тактика ведення в неонатальному періоді:
При задніх уретральних клапанах у новонароджених для відновлення прохідності уретри в постнатальному періоді встановлюється катетер або проводиться везікостомія. Після корекції водного і електролітного обмінних порушень і стабілізації ниркової функції здійснюють видалення або електрокоагуляцію задніх уретральних клапанів. Навіть у випадках успішної терапії діти помирають в перші тижні від легеневої недостатності. Крім того, у багатьох дітей, які вижили, спостерігається неадекватна функція нирок, яка може прогресувати і потребувати діалізу і трансплантації.
Література:
- Warkany J. Congenital Malformations: Notes and Comments. Year Book Publisher. Chicago. 1981:1081-1082.
- Ромеро Р, Пилу Дж. Пренатальная диагностика вродженных пороков развития плода.- М.: Медицина, 1994.- С. 281-290.
Переглянуто редакційною колегією I.B.I.S.: 05/02/2002
Життя з синдромом Лоу
(Lowe Syndrome)
Л.С. Євтушок
Зав. медико-генетичною консультацією
Рівненського клінічного лікувально-діагностичного центру
О.О. Михасюк
Спеціаліст з інформаційного забезпечення
Українсько-Американської Програми запобігання вродженим вадам розвитку
Вперше синдром Лоу був описаний в 1952 докторами Лоу, Терреєм та МакЛахланом. Вони описали три дитини чоловічої статі, що мали однакові клінічні прояви та хвороби, які до цього часу не асоціювались одна з іншою. Оскільки вони не могли з’ясувати причину такого порушення, випадкам поєднання цих симптомів і характерних ознак було надано статусу “синдрому”.
В медичній літературі хвороба стала відомою під назвою “синдром Лоу”, на честь доктора Чарльза Лоу, керівника групи вчених, що описали дане порушення. Цей синдром також є відомим як “окуло-церебро-ренальний синдром”, що проявляється на трьох основних системах органів – очі, мозок та нирки.
В наступні роки лікарі з’ясували, що синдром Лоу є спадковою хворобою, що вражає лише чоловіків. Вона спричиняється поодиноким пошкодженим геном в Х-хромосомі, одній з двох статевизначающих хромосом. Зазвичай, цей ген виробляє спеціальні ензими, що беруть участь в метаболізмі інозиту (вітаміну В8). Але ж оскільки даний ген має дефект, необхідний ензим не виробляється. Тому можна із певністю сказати, що синдром Лоу спричиняється саме нестачею даного ензиму.
Синдром Лоу є досить рідкісним захворюванням. Випадки цього порушення були зафіксовані в усьому світі і мали приблизно однакову частоту трапляння в усіх етничних групах. Ніхто не знає напевно, скільки чоловік має синдром Лоу. В Сполучених Штатах частота випадків синдрому Лоу становить від 1 до 10 на 1000000 підлітків-хлопчиків, або від 200 до 2000 існуючих випадків.
Медичні ознаки:
Діагноз
Діагноз “синдром Лоу” можна ставити коли в новонародженої дитини спостерігається вроджена катаракта, дисфункція нирок, та характерні прояви порушень нервової системи в новонароджених хлопчиків, особливо якщо в родинній історії спостерігались випадки Х-наслідування. Зазвичай, синдром Лоу діагностується лікарем-генетиком, однак і інші лікарі також можуть ставити такий діагноз.
Однак в ранньому дитинстві діагноз може не завжди бути чітким та незаперечним. Хоча катаракта та зниження м’язового тонусу (гіпотонія) визначаються одразу після народження, інші прояви порушень зору та нервової системи можуть проявитися пізніше. Лабораторні дослідження щодо порушення функції нирок також можуть не дати результатів одразу після народження. Слід зауважити, що ступінь враження нирок, нервової системи та інших органів може бути різною в кожного конкретного пацієнта.
Для остаточної діагностики синдрому Лоу необхідно зробити аналізи на наявність або відсутність специфічного ензиму в клітинах шкіри. Під час даної процедури в пацієнта береться маленький зріз шкіри і посилається в спеціалізовану біохімічну лабораторію для проведення аналізів.
Факт нестачі в клітинах шкіри ензиму, що викликає синдром Лоу є єдиною стовідсотковою гарантією діагнозу. Тепер вражені діти виявляються протягом перших місяців життя, оскільки дослідження синдрому Лоу тривають і з’являються нові тести для його діагностування.
Очі
- Катаракта – в усіх вражених чоловіків катаракта проявляється одразу після народження. Часом її не діагностують впродовж перших тижнів життя, однак вона завжди є. Катаракта – помутніння або затемнення кришталика ока. Зазвичай, кришталики – як лінзи в камери, вони мають бути чистими і прозорими. Лінзи фокусують світло на сітчатку що знаходиться на задній частині ока, займаючи 2/3 його розмірів. Сітчатка ж працює подібно до фотоплівки – “знімає картинку” та передає її через оптичний нерв в мозок. Так відбувається процес бачення. Замутнені або молочно-бліді кришталики не дають світлу проходити скрізь око і робити чітке фокусування картинки. Катаракту слід видалити якомога раніше. Найкраще це робити протягом перших днів життя, оскільки отримання чіткого зображення предметів навколишнього середовища є дуже важливим для мозку дитини, що розвивається. Сіро-білі кришталики видаляються хірургічним шляхом із застосуванням інструментів для мікрохірургії ока при загальній анестезії. Однак після видалення помутнілих кришталиків око матиме зменшену здатність до фокусування. Окуляри або контактні лінзи компенсують таке порушення зору. Для більшості дітей своєчасне видалення помутнілого кришталика та корегуюча терапія із застосуванням окулярів дає гарантію значного покращання зору. При цьому лікарі не радять застосовувати штучні пластикові лінзи-протези.
- Глаукома – глаукома розвивається в кожного другого пацієнта із синдромом Лоу. Це хвороба, при якій внутрішньоочний тиск підвищується до такого високого рівня, що пошкоджує оптичний нерв та зір в цілому. Підліткова глаукома може мати згубні наслідки. Вона проявляється або одразу після народження, або протягом першого року життя. Оскільки око дитини має малі розміри, а його тканина більш еластична, ніж у дорослої людини, зростаючий неконтрольований тиск протягом певного часу може призвести до ненормального збільшення розмірів ока. Цей симптом має назву буфтальм, або “биче око”. Підліткову глаукому важко лікувати, до того ж не існує єдиного визнаного метода її лікування. Очні краплі можуть знизити тиск, однак самі по собі мало ефективні. Якщо крапельна терапія не дає позитивного результату, застосовують хірургічне втручання – в середині ока створюють новий канал для очної рідини, щоб вона могла легше витікати, не створюючи надлишкового тиску. Інколи встановлюють штучний клапан на задню частину ока для того, щоб контролювати рівень і тиск очної рідини. На жаль, в деяких випадках глаукома є настільки важкою, що не піддається лікуванню. В таких випадках зір втрачається назавжди. Загроза глаукоми є основною підставою для проведення огляду очей новонародженої дитини одразу після пологів. І, хоча ризик глаукоми зменшується після першого року життя, пацієнти із синдромом Лоу мають принаймні раз на рік проходити обстеження у окуліста.
- Дегенерація сітківки – сітчатка є прозорим “склом” на передній поверхні ока. З невідомих причин в декого з пацієнтів із синдромом Лоу, навіть тих, що не мають глаукому, виникає рубцева тканина на сітківці, що називається келоїд. Правда, дехто з лікарів вважає, що це новоутворення не є справжнім келоїдом, а лише фібромою або доброякісною фіброзною пухлиною. Келоїди можуть спричиняти порушення зору, коли вони вкривають сітківку і не пропускають світло. Келоїди можуть вражати як одне, так і обидва ока. Частота виникнення келоїдів невідома, але є дані, що свідчать про те, що кожен другий хворий із синдромом Лоу віком від 13 до 19 років страждає на дегенерацію сітчатки. Своєчасне лікування келоїдів може дати непоганий результат. Їх можна лікувати хірургічно, медикаментозно або із застосуванням радіологічної терапії. Трансплантація сітчатки дає погані результати в дітей із синдромом Лоу. Часто прогресуючий розвиток келоїдів призводить до сліпоти.
- Косоокість – багато дітей із синдромом Лоу мають косоокість. В них очі не можуть рухатись одночасно і зосереджуватись на предметах, що знаходяться прямо перед ними. Зазвичай, при цьому одне око відхиляється в сторону. В дітей із вродженою косоокістю зір погіршується не лише через неї, але й тому, що мозок отримує неповну і неточну інформацію про об’єкти навколишнього світу і не може реагувати відповідно. Лікування косоокості слід починати з ретельного обстеження рухливості очей, їх м’язів та рефракції. Згідно з результати обстеження призначаються окуляри або контактні лінзи. Зазвичай це робиться тоді, коли обидва ока не можуть зосереджуватися на предметах попереду.
- Ністагм – даний термін означає неконтрольовані мимовільні рухи очей, вони не затримуються на жодному предметі ні за яких обставин. Ністагм може спричинятися багатьма причинами, кожна з яких пов’язана із погіршенням зору. Часом, затримка розвитку мозку може спричиняти порушення координації очей. Сам по собі ністагм не призводить до втрати зору, однак має неприємний зовнішній ефект.
На жаль, для ністагму немає лікування, крім можливості провести корекцію зору якомога швидше. При синдромі Лоу погіршення зору може починатися з вродженої катаракти і хірурги-офтальмологи можуть своєчасно провести операцію і зберегти зір дитині. Також при синдромі Лоу вражається райдужна оболонка ока.
Мозок
Нервова система включає в себе головний мозок, спинний мозок, нерви та м’язи. Синдром Лоу спричиняє порушення в усіх цих органах. Проблеми, пов’язані із порушенням мозку – розумове відставання, корчі, проблеми з поведінкою, фізичні зміни в мозку, гіпотонія.
- Розумове відставання – в пацієнтів із синдромом Лоу спостерігаються різні ступені розумового відставання. Приблизно 10-25% пацієнтів мають відносно нормальний розумовий розвиток, ще 25% – легкий та середній ступені розумового відставання. Решта пацієнтів мають важку затримку розумового розвитку. При народжені неможливо встановити, наскільки важким буде розумове відставання. Розумовий розвиток формується протягом життя.
- Корчі – спостерігаються в половини усіх пацієнтів. Молодші діти можуть мати корчі на фоні високої температури. Зазвичай, вони не вимагають медикаментозного лікування. В більш дорослих хлопчиків корчі мають інший – основно-моторний характер. Корчі цього типу лікуються різними антиконвульсивними засобами. Таких пацієнтів можна відмічати під час проведення електроенцефалограми (ЕЕГ), яка обов’язково покаже аномалії мозку і допоможе виявити ту ділянку мозку, в який виникають корчі.
- Проблеми з поведінкою – пацієнти із синдромом Лоу зазвичай дуже життєрадісні, комунікабельні та милі, однак можуть мати особливі риси поведінки, що можуть порушувати розмірений режим дня. Сюди відносяться раптові спалаху гніву, загальмованість, незвичні рухи, що повторюються, нездатність до концентрації уваги, надмірна зосередженість на чомусь. В деяких випадках пацієнти можуть проявляти агресію та самонівечення. Ці ознаки поведінки є типовими при синдромі Лоу. Результати деяких досліджень свідчать про те, що основні проблеми із поведінкою виникають в пацієнтів у віці 8-13 років. Часом важкі проблеми із поведінкою зберігаються до підліткового віку. Тут на допомогу приходять методики коригування поведінки, а інколи навіть медикаментозне лікування.
- Фізичні зміни головного мозку – знімки мозку, отримані при магнітно-резонансному скануванні можуть показати аномальні сигнали в мозку. Ці аномалії спричиняються тонкими кистозними утвореннями, наповненими рідиною, що утворюються протягом першого року життя. Вони не ростуть і не заважають нормальному функціонуванню мозку. Ці новоутворення є скоріше медичною загадкою, аніж проблемою. Точна причина їх виникнення невідома.
- Гіпотонія – гіпотонія або слабкий м’язовий тонус відмічається в усіх пацієнтів із синдромом Лоу. Вона може проявлятися як одразу після народження, так і протягом перших місяців життя. Гіпотонія проявляється в надмірній гнучкості дитини та малій м’язовій силі. В ранньому дитинстві через гіпотонію можуть виникати проблеми із триманням голови та годуванням (через ослаблене смоктання). Коли хлопчики підростають, в них спостерігається затримка моторного розвитку. Наприклад, понад 25% хлопчиків із синдромом Лоу починають ходити у віці 3-6 років. Ще 75% починають ходити у віці 6-13 років. З віком гіпотонія дещо зменшується, однак більшість пацієнтів ніколи не досягають нормального рівня фізичного розвитку, через що виникають відповідні проблеми коли вони дорослішають. Ослаблені або гіпермобільні суглоби є типовою ознакою зниженого м’язового тонусу. В половини хлопчиків розвивається сколіоз або викривлення спини через підвищену слабкість м’язів спини. Підвищений ризик розвитку сколіозу спостерігається у віці 13-18 років. Також протягом усього життя є загроза утворення кил через слабкі м’язи живота і черевної порожнини. Гіпотонія при синдромі Лоу є первинною по відношенню до дисфункціїї нервової системи. В хворих спостерігається знижений вміст м’язового ензиму, що називається креатинін кіназа (КК). Спеціальні м’язові проби такі, як електроміографія або м’язова біопсія є не обов’язковими при синдромі Лоу. Глибокі сухожилкові реакції (наприклад, колінний рефлекс) зазвичай, є відсутніми одразу після народження. Це може бути спричиненим як пошкодженням нервової системи, так і дисфункцією спинного мозку. Відсутність цих рефлексів не створює перешкод для нормального функціонування. Найкращі результати при лікуванні гіпотонії дає терапія, що починається з раннього дитинства.
Нирки
Клінічні ознаки аномалій нирок можуть бути відсутніми одразу після народження, однак завжди проявляються протягом першого року життя. Головна проблема із нирками при синдромі Лоу полягає у відсутності певних речовин у сечі, таких як бікарбонат, амінокислоти, фосфати та L-каротини. Це відбувається через пошкодження ниркових канальців, які мають фільтрувати кров від шлаків. В здорових людей вищеперелічені речовини спочатку виділяються, а потім повторно всмоктуються. При синдромі Лоу ці речовини виділяються нирками, але повторно не абсорбуються. Ця аномалія відома як дисфункція ниркових канальців типу Фанконі і може спостерігатися при деяких інших синдромах.
Протягом перших 10-12 місяців життя в дитини із синдромом Лоу може не спостерігатися негативних змін в хімічному складі крові та сечі. Тому лікарі, які скептично ставились до попереднього діагнозу “синдром Лоу”, можуть відкинути його через те, що довгий час аналізи не показують характерних змін в крові та сечі.
Деякі речовини, які виділяються разом із сечею, є необхідними для нормального протікання біохімічних процесів і їх відсутність може призвести до серйозних змін в метаболізмі організму. Хворі діти можуть мати різноманітні прояви порушення функції нирок. Більшість з них потребує замісної терапії, коли нестача необхідних речовин компенсується прийомом необхідних медикаментів. Тип та дозування ліків має бути визначено індивідуально для кожного пацієнта.
В разі проведення замісної терапії слід робити аналізи крові та сечі через певні проміжки часу для того, щоб простежити ефективність лікування. Крім того, оскільки в хворих нирки не можуть нормально утримувати воду та концентрувати сечу, їм необхідно вживати більше рідини для того, щоб запобігти дегідратації.
По досягненні хворими 10-річного віку починають розвиватися інші порушення нирок, що часто призводить до повної ниркової недостатності відмови десь у 40 років.
- Недостатність бікарбонатів – низький рівень бікарбонатів в крові проявляється в порушенні кислотно-лужного балансу і має назву “ацидоз”. Характерною ознакою цього стану є зниженням рівня рН тіла. Клінічні симптоми ацидозу включають летаргію, зниження апетиту, відставання у рості. Якщо такий стан залишити без лікування, важка форма ацидозу може становити загрозу для життя пацієнта. 2/3 хворих із синдромом Лоу страждають на довготривалий ацидоз і потребують замісної терапії. При цьому можна вживати препарати, що містять бікарбонати або перетворюються на бікарбонати в процесі біохімічних процесів, такі як цитрат.
- Недостатність калію – втрата калію з сечею – гіпокаліємія або зниження рівня калію в крові. До симптомів гіпокаліємії відносяться м’язова слабкість, підвищена втома, надмірне сечовиділення, спрага. Зазвичай, в таких випадках застосовують замісну терапію.
- Недостатність амінокислот – в більшості випадків підвищене виділення амінокислот з сечею не спричиняє медичних проблем та не потребує лікування. Однак, цей симптом є корисним для визначення причини аномальної роботи нирок.
- Недостатність фосфатів – втрата фосфатів з сечею хворого може призвести до розвитку гіпофосфатемії (зниженню рівня фосфатів в крові). Такий патологічний стан спостерігається в 1/3 усіх хворих із синдромом Лоу. Як наслідок, можуть виникнути серйозні проблеми із кістками, такі як рахіт або “м’які” кості. Для лікування застосовують пероральні фосфатні додатки.
- Недостатність L-каротину – L-каротин є маленькою молекулою протеїну, яку потребують наші клітини для перетворення жирів на енергію. Зазвичай такий стан асоціюється із постійним відчуттям голоду, що супроводжується характерними для гіпотонії симптомами, збільшенням або порушенням роботи печінки, низьким рівнем цукру в крові, змінами в самопочутті. Деякі пацієнти при цьому потребують замісної терапії.
Кістки
- Рахіт та “м’які” кістки – в багатьох дітей із синдромом Лоу спостерігаються, так званий, ефект “м’яких кісток”, рахіт або часті переломи кісток. Це є результатом цілого комплексу причин. В процесі свого розвитку кості мають затвердіти. Фосфати, кальцій, вітамін D та інші речовини і гормони у поєднанні із нормальним кислотним балансом – все це є неодмінними складниками процесу кісткоутворення та їх щільності. Для підтримання та розвитку кісток необхідні фізичні вправи, що збільшують м’язову масу біля кісток, яка, в свою чергу, оберігає кості від переломів. Низький рівень фосфатів у крові, ацидоз, гіпотонія, порушення функцій нирок – усе це може стати причиною виникнення порушень розвитку кісток, рахіту або остеомаляції. Рахіт або остеомаляцію можна попередити, якщо приймати нейтральні фосфатні добавки у поєднанні із калієвими та бікарбонатними препаратами. Побічним ефектом фосфатотерапії може бути пронос, тому дозу слід визначати дуже обережно. Також слід призначати профілактичний прийом вітаміну D. Усі медичні препарати та їх дозування мають бути призначеними лікарем, що веде спостереження за даним пацієнтом. Періодично слід робити рентгенівські знімки кісток та здавати аналізи крові і сечі. Зміна ліків має бути узгоджена із лікарем для того, щоб запобігти погіршенню стану.
- Переломи – приблизно 5-% пацієнтів мають переломи. При цьому часто страждають ноги (особливо під час навчання ходити). 1/3 пацієнтів мають неодноразові переломи кінцівок.
- Коротка статура – майже усі новонароджені із синдромом Лоу мають нормальну довжину при народженні, однак по досягнені віку 1 року вони починають відставати у зрості від своїх однолітків. Ріст продовжується, однак набагато повільніше. Зараз тривають дослідження щодо ефективності та безпеки лікування гормонами росту.
- Набряк суглобів та артрит – ці та інші ортопедичні проблеми виникають в пацієнтів із синдромом Лоу, починаючи з підліткового віку. В деяких випадках ці проблеми можуть набувати дуже складних форм. Причина виникнення даної патології невідома, отож не існує і ефективного лікування крім симптоматичного, спрямованого на полегшення болю.
- Зуби – багато дітей із синдромом Лоу потребують частого лікування зубів. Високе піднебіння, зменшений ротовий отвір, рахіт та інші фактори часто спричиняють зміни зубів: неправильне їх розташування, зниження вмісту кальцію, затримка появи перших зубів. Регулярне відвідування стоматолога слід починати одразу після появи першого молочного зуба.
Спеціальні поради щодо профілактики та лікування хвороб у пацієнтів із синдромом Лоу:
Деякі звичайні хвороби пацієнтів можуть потребувати особливого підходу:
- Умови порушення метаболічного балансу – будь-яке порушення встановленого денного режиму прийому рідини та ліків може спровокувати виникнення важкого порушення метаболічного балансу. Часто такий стан виникає внаслідок інфекційних хвороб (застуди, грипу) або блювання чи проносу. В таких випадках батькам слід уважніше спостерігати за хворою дитиною і в разі потреби негайно інформувати лікаря, оскільки може знадобитися внутрішньовенне введення рідини та інших медикаментів. Крім того, пацієнтам із синдромом Лоу слід призначати профілактичні курси внутрішньовенного введення рідини під час проведення операцій, анестезії або інших процедур, при яких вимагається тимчасове припинення вживання їжі та рідини через рот.
- Респіраторні захворювання – в дітей із синдромом Лоу спостерігається підвищена схильність до виникнення пневмоній внаслідок гіпотонії (слабке відкашлювання). Під час респіраторних захворювань вони мають знаходитись під наглядом лікаря. Батьки можуть навчитися самостійно виконувати постуральний дренаж, що може полегшити відкашлювання харкотиння.
- Закреп – кожний другий пацієнт має проблеми із дефекацією. Це можна пояснити зниженням м’язового тонусу. Хоча деякі лікарі вважають, що постійний закреп є характерною ознакою синдрому Лоу – прямий ефект впливу ураженого гену на м’язи кишечника та їх нездатність сприяти пересуванню калових мас вздовж кишечника. Лікування полягає у вживанні великої кількості клітковини та рідини. В разі неефективності дієтлікування слід приймати медичні препарати. Однак це слід робити лише під наглядом лікаря. При цьому НЕ РАДЯТЬ застосовувати клізми, оскільки вони можуть викликати серйозні порушення метаболічного балансу і становити загрозу для життя хворого.
- Кісти – виникнення кіст є загальною проблемою при синдромі Лоу. Вони можуть виникати в різних органах, включаючи ротову порожнину, нирки, мозок та шкіру. Частота їх виникнення невідома. Блакитна склера часто виникає в ротовій порожнині дітей, однак зазвичай зникає у підлітків після формування корінних зубів. Проте в старших хлопчиків виникають кісти в нирках та на шкірі. В підлітків часто виникають кісти на шкірі в нижній частині спини та на сідницях. Вони можуть бути дуже болісними, до них часто потрапляє інфекція. Причина виникнення кіст невідома, однак скоріше за все це є невід’ємною складовою комплексу синдрому Лоу. Можна проводити курси лікування препаратами ретинової кислоти, однак її вплив на нирки при синдромі Лоу невідомий.
- Неопущення яєчка – неопущення яєчка виникає в кожного третього хворого. При цьому в третини хворих з часом яєчки самостійно опускаються на місце. Решта хворих потребує постійного нагляду для визначення необхідності проведення хірургічної корекції.
Прогноз:
Ступінь вираженості проявів ураження в пацієнтів із синдромом Лоу є дуже різним. Прогноз, яке майбутнє чекає на того чи іншого пацієнта дати неможливо, однак можна спиратися на так званий “типовий шлях перебігу хвороби”. Перша (неонатальна) фаза починається одразу після народження із визначенням катаракти та низького м’язового тонусу. Ця фаза характеризується хірургічним видаленням катаракти та післяопераційним доглядом за дитиною. Саме в цей час ставиться діагноз “синдром Лоу”.
Друга фаза починається з першого року життя, коли виникають перші проблеми із функціонуванням нирок. Ця фаза може тривати як кілька місяців, так і 5-8 років. В цей період необхідні часті консультації лікаря для призначення нових препаратів для замісної терапії, проведення лабораторних аналізів крові та сечі.
Третя фаза починається в старшому дитинстві або ранньому підлітковому віці. В цей час спостерігаються різноманітні порушення функції нирок, суглобів, проблеми зі шкірою та поведінкою. Нирки продовжують вимивати з організму бікарбонати і тому потреба в замісних препаратах зростає. Порушення роботи нирок можуть становити загрозу для життя. Набряки суглобів спричиняють сильний біль та обмежують рухливість кінцівок. Завдяки інтенсивному лікуванню більшості цих симптомів можна запобігти і, таким чином, продовжити тривалість життя до 20-30 років, а часом, навіть, до 40 років. Смертність, що спричинена ускладненнями при синдромом Лоу (інфекційні захворювання, дегідратація, пневмонія), відмічається в різному віці. Усі ці чинники щільно досліджуються і є надія досягти значних успіхів у лікуванні якщо не самого синдрому Лоу, то, принаймні, його ускладнень.
Генетичні особливості:
Синдром Лоу є генетичним захворюванням, що спричиняється змінами в гені. В багатьох родинах синдром Лоу є спадковим, що передається від матері-носія до її ураженого сина; інші родичі матері також можуть мати ген-мутант. В інших родинах хворий хлопчик є першим ураженим. Родини, що мають хворих із синдромом Лоу мають чітко визначити умови успадкування даної хвороби для того, щоб знати, хто ще має ризик виникнення даної патології. Це допоможе встановити загрозу повторного народження хворої дитини та відповідно до цього спланувати життя родини.
A. Як успадковується синдром Лоу.
- Гени – синдром Лоу є “вродженим порушенням метаболізму”. Метаболізм є складною системою фізичних та хімічних процесів, що відбуваються в кожній ділянці людського тіла. Усі ці процеси контролюються генами, що є основою життя і які усі ми успадковуємо від наших батьків. Коли в одному з них відбувається мутація (зміна або перетворення), це спричиняє “помилку” в метаболізмі, спричиняючи хворобу. Більшість мутацій в генах виникає спонтанно. Однак, коли вони вже виникли, вони є зафіксованими в генетичній структурі людини. Якщо хворий пацієнт виживає, він чи вона можуть передати цей уражений ген своїм нащадкам. Таким чином хвороба стає спадковою і передається від одного до іншого члена родини.
- Хромосоми – це є гени, що згруповані разом. За виключенням червоних кров’яних тілець та репродуктивних клітин, кожна людська клітина містить 23 пари хромосом, всього 46 хромосом. Один член пари береться від матері, другий від батька. Репродуктивні клітини, яйцеклітина та сперматозоїд, кожен з них містить 23 хромосоми – по одному представнику кожної пари. В момент запліднення сперматозоїд зливається із яйцекліткою, утворюючи клітину із 23 парами хромосом та повним набором генетичного матеріалу для розвитку та формування нової людини. 22 пари хромосом містять у собі гени, що визначають основні характеристики тіла, такі як колір очей, статура, зріст. Ще одна пара відрізняється від інших – вона визначає стать майбутньої людини. Ця пара має “X” та “Y” хромосоми. Жінки отримують дві однакових X-хромосоми від матері та від батька. Чоловіки мають “X” хромосому від матері та “Y” від батька. Сперматозоїд чоловіка передає або “X”, або “Y” хромосому, однак в жодному разі не обидві одразу. Якщо через батьківський сперматозоїд передається Х хромосома, то дитина буде жіночої статі (ХХ), якщо ж Y, тоді народиться хлопчик (XY).
- Успадкування по Х-ланці. З початку 60-х років вчені знали, що ген, який спричиняє синдром Лоу знаходиться в Х-хромосомі. Хвороби, які спричиняються порушеннями генів Х-хромосоми називаються “Х-зчепленими” і мають єдиний спосіб передачі. Зазвичай, страждають від цих хвороб чоловіки, хоча носієм завжди є жінка. В жінок-носіїв синдрому Лоу одна Х-хромосома є нормальною, а друга уражена. Чоловік, що успадковує свою єдину Х-хромосому від матері і може успадкувати її Х-хромосому із синдромом Лоу, буде ураженим цим синдромом оскільки лише Х-хромосома містить його генетичну інформацію. В Y-хромосомі її немає.
- Носії-жінки, що мають ген синдрому Лоу в їх Х-хромосомі називаються носіями, тому що вони зберігають інформацію про хворобу всередині. Із кожною вагітністю така жінка має 50% шансів передати уражену хромосому своїй дитині, якщо це буде хлопчик. Якщо ж дитина буде жіночої статі вона також має 50% ризику отримати від матері хворий ген і відтоді бути його носієм. Отож, жінка-носій має шанси 1 до 4 мати дитину, уражену синдромом Лоу, якщо це буде хлопчик. Один шанс з чотирьох мати нормального хлопчика; 1:4 що її дочка буде носієм хворого гена і 1:4 що вона не буде його мати.
Б. Генетичне консультування.
Пари, що мають ризик мати дитину із синдромом Лоу, повинні планувати народження дитини під наглядом лікаря-генетика. Генетики зможуть вказати на ступінь ризику народити хвору дитину та визначити, чи дійсно жінка є носієм даного гена.
- Визначення носія. Родини, що відносяться до групи ризику по синдрому Лоу, за бажанням можуть визначити вірогідність народження хворої дитини. До жінок, що є потенційними носіями, відносяться матері та сестри ураженого хлопчика, його тітки по материнській лінії та їх дочки. Для того, щоб визначити, чи є жінка носієм хворого гена, важливо оглянути усіх членів родини, що вже мають або планують мати дітей. Генетики та генетичні консультації можуть визначити ризик для таких пар мати дитину із синдромом Лоу. Інколи статус жінки-носія можна встановити з родинної історії хвороби. Наприклад, мати дитини, ураженої синдромом Лоу з великою вірогідністю буде носієм цього гена, особливо, якщо в попередній історії родини вже були випадки народження хворих дітей. Однак, якщо до цього не було зафіксовано жодного випадку народження дітей із синдромом Лоу в родині, мати може і не бути носієм хворого гена, а порушення, що спричинило ураження її дитини було викликано новими мутаціями в його власних генах. В будь-якому випадку, мати має бути обстежена для остаточного визначення її статусу носія. Це допоможе встановити причину хвороби та ризик для майбутніх дітей. В більшості випадків статус носія можна визначити при звичайному огляді. Майже усі жінки-носії гена синдрому Лоу мають незначні зміни в кришталику ока, особливо це помітно у підлітковому віці. Ці зміни проявляються у вигляді маленьких точок та вузликів на кришталику ока, що не порушують гостроти зору. Отож, в своєму висновку лікар-генетик має спиратися на дані попереднього огляду офтальмолога. Проте, коли мова йде про дівчат-підлітків, остаточний висновок про статус носія слід робити не раніше досягнення ними 15 річного віку. Також слід пам’ятати, що були зафіксовані випадки, коли жінка була носієм ураженого гена, але не мала змін в кришталику ока.
- Підходи до планування сім’ї. Сімейні пари, що відносяться до групи ризику, мають зробити вибір після того, як було встановлено носія гена. Дехто покладається на долю і не застосовує методів контрацепції для запобігання небажаної вагітності. Дехто відмовляється від ідеї мати власних дітей і всиновлює одну чи навіть більше дітей. Інші намагаються народити дитину, використовуючи донорську яйцеклітину, що не має загрози виникнення спадкової патології. Є й такі, що намагаються народити дитину жіночої статі, оскільки вона не буде уражена синдромом Лоу, навіть, якщо і буде носієм. Під час проведення пренатальних аналізів можна точно визначити, якої статі буде малюк. В разі ймовірності народження дитини чоловічої статі у жінки, що відноситься до групи ризику, проводяться спеціальні ензимні дослідження для визначення того, чи є плід ураженим. Цей аналіз можна провести для підтвердження навіть, якщо попередній генний аналіз не виявив наявності ураженого гена. Оскільки було встановлено, що синдром Лоу викликається порушенням гена, варто провести спеціальні ДНК-дослідження для його визначення. Нещодавно для подружніх пар з групи ризику була запропонована нова технологія, що називається “преімплантаційна генетична діагностика”. Вона полягає у проведення штучного запліднення в пробірці із наступним вивченням ДНК заплідненої яйцеклітини на наявність гена, що спричиняє хворобу. В разі відсутності хвороби, цей зародок може бути імплантованим матері. Дана методика була з успіхом застосована для попередження цілої низки спадкових хвороб. Однак, ситуація із синдромом Лоу в даному випадку є ускладненою через те, що неможливо провести ензимні проби в зародка, оскільки матеріалу для дослідження надто мало. Є багато факторів (внутришньосімейні, етичні, фінансові, релігійні), що впливають на рішення, яке має прийняти кожна родина. Немає універсальної “правильної” відповіді і кожне подружжя має саме визначити своє майбутнє та майбтнє їх дитини.
В. Дослідження.
Протягом перших 30 років після відкриття синдрому Лоу, дослідники зробили багато для усвідомлення його причин та наслідків. В 1954 році була описана особлива форма порушення роботи нирок, що пов’язана із даним синдромом. Успадкування по Х-ланці було визначено в 1957 році. А можливість визначати жінок-носіїв з’явилася в 1976 році.
Дослідження синдрому Лоу ускладнюється ще й тим, що дана патологія є досить рідкісною і вченим бракує матеріалу для дослідження. Ця ситуація дещо змінилася з 1983 року, коли була створена Асоціація Хворих із Синдромом Лоу (АХСЛ). Відтоді у вчених з’явилося багато добровольців, готових повністю присвятити себе дослідженням свого захворювання заради здоров’я інших. Інша проблема полягає в тому, що дана хвороба притаманна лише людям і не має жодної, принаймні подібної, моделі в тваринному світі.
Великим досягненням у вивченні синдрому Лоу стало відкриття у 1986 році докторами Льюісом та Нуссбаумом локалізації порушеного гену в довгому плечі Х-хромосоми. Це дало поштовх для точного встановлення і самого гену-порушника, що сталося в 1992 році. Було з’ясовано, що хвороба спричиняється нестачею ензиму фосфатіділіноситолу 4,5-біфосфат 5 фосфат, що є залученим до нормального перебігу метаболічних процесів. Оскільки даного ензиму не вистачає організму, що розвивається, в клітинах відбуваються певні порушення, які, в свою чергу, спричиняють порушення роботи нирок, мозку та катаракти.
Г. Розвиток та навчання.
Не всі пацієнти із синдромом Лоу подібні один до одного. Наявність та ступінь важкості симптомів дуже мінлива, навіть в межах однієї родини. Як наслідок, хворі діти в різному віці оволодівають певним набором необхідних вмінь та навичок. Однак, слід відмітити, що хлопчики із синдромом Лоу потребують посиленого навчання та розвитку. Це дає шанс розвинути їх до досить належного рівня.
Через фізичні та розумові обмеження хлопчики із синдромом Лоу часто відстають від своїх однолітків. Зазвичай, їх розвиток відбувається за звичайною схемою, хоча в деяких галузях (наприклад, мова) може значно випереджувати інші. Існує велика кількість різноманітних методик, спрямованих на розвиток як розумових так і прикладних навичок.
Майже усі пацієнти із синдромом Лоу є дуже комунікабельними та життєрадісними, мають чудове відчуття гумору. В багатьох є неабиякі здатності до музики. Коли вони себе нормально почувають та знаходяться в приємному середовищі, вони щасливі та веселі. Вони потребують багато зусиль та терпіння, але при цьому можуть стати функціонально повноцінними членами своєї родини, що мають власну індивідуальність.
- Раннє дитинство.
Дослідження свідчать, що лікування та тренування хворих слід починати з самого раннього дитинства. Своєчасно розпочаті заняття дозволять дитині швидше вчитися та значно зменшать рівень розумового та фізичного відставання.
В процесі розвитку дитини чи не найважливішу роль відіграють батьки. Саме з них починається цей розвиток і на них лежить відповідальність за дитину.
Фізичний розвиток дитини є зниженим, але багато в чому піддається лікуванню та виправленню. Зазвичай, батьки проходять спеціальну підготовку під наглядом лікаря для того, щоб в разі необхідності вміти самостійно надати допомогу своїй дитині. В ранньому віці дуже корисними є вправи для м’язів шиї, тренування та масаж ніг і спини. Не слід чекати швидкого прогресу – це дуже довгий і складний процес, однак він завжди приносить свої плоди.
Розвиток мовлення характеризується його затримкою. Це можна пояснити, як фізичними особливостями (зниження м’язового тонусу, порушення зору, високе піднебіння), так і затримкою розумового розвитку, що призводить до нерозуміння значення слова, нездатності правильно сприймати чи передавати інформацію. Хворі діти починають імітувати звуки десь у віці 2-2,5 років. До семирічного віку майже усі вони вміють розмовляти.
Проблеми із годуванням. В деяких випадках ті самі фактори, що спричиняють затримку мовлення впливають і на порушення прийому їжі. Слабкі м’язи спричиняють погану координацію рухів язика, губ та щелеп, які необхідні для нормального жування та ковтання. Результатом цього є важкий перехід дітей від дитячого до нормального харчування. Стимулююча терапія дозволяє дітям із синдромом Лоу перейти на нормальну їжу десь у віці 5 років.
- Початкова та середня школа.
Шкільна програма для дітей із синдромом Лоу залежить від рівня розвитку особистості та можливостей школи. Дехто з хлопчиків відвідує класи для дітей із затримкою розумового розвитку. Проте дехто може відвідувати і нормальні класи. Діти із значним розумовим відставанням виховуються під наглядом батьків та спеціальних вчителів.
Ходіння. Терапевтичне лікування, що триває до цього часу, має особливу концентрацію на розвиток самостійної ходи. У віці від 5 до 13 років понад 70% пацієнтів вже можуть самостійно ходити.
Самообслуговування та особиста гігієна. Навчання самостійно ходити до туалету має велике значення для хлопчиків. Починаючи тренування з раннього дитинства, можна досягти певних результатів і у віці 5-13 років майже усі хворі можуть самостійно себе обслуговувати. Крім того, проблеми можуть виникати і з іншими сферами самообслуговування, такими, як одягання, вмивання і таке інше. В багатьох хлопчиків виникають проблеми із натяганням шкарпеток, застібанням гудзиків, писанням.
Статевий розвиток. Зазвичай, статевий розвиток у хворих на синдром Лоу затриманий, однак розвивається за нормальною схемою.
Поведінка. Як було зазначено вище, в декого з хлопчиків у віці 5-13 років виникають серйозні проблеми із поведінкою. Отож, це вимагає від батьків та вчителів додаткових зусиль на вгамування цих проблем. Емоційні зриви порушують встановлений процес навчання, соціальну адаптацію, життя в родині. В цей час діти та їх батьки потребують консультування у психолога, для визначення ефективних методів уникання конфліктів та перемикання уваги та емоційної скерованості дитини на щось інше, наприклад, навчання.
- Підлітковий вік.
Протягом усього життя більшість хлопчиків із синдромом Лоу живуть вдома із своїми батьками. Проте є випадки, коли родина приймала рішення помістити дитину в спеціалізований заклад. Це пояснювалось різними причинами – необхідністю посиленого медичного догляду, потребою в спеціальному навчанні, серйозними порушеннями соціальної поведінки. В більш дорослому віці деякі хворі можуть перейти до спеціальних родинних груп. Дехто з них працює на допоміжних роботах або в спеціальних програмах для людей із розумовим чи фізичним відставанням.
Д. Вплив на родину.
Мабуть, немає жодної людини, яка б була повністю готова до росту та виховання дитини, не кажучи вже про дитину із вродженою вадою. Перед батьками встає велика кількість проблем і треба навчитися жити з ними. Їм доводиться миритися із неповноцінністю сина, жахливими діагнозами, що йому ставлять лікарі. Їм доводиться долати медичну та освітню бюрократію. Вони мають набратися терпіння і чекати, коли їх турбота дасть плоди. Крім того, вони мають також якось впоратись із власним відчуттям відчаю, розчарування, страху за майбутнє.
Усе починається з моменту, коли батьки усвідомлюють, що з їх дитиною “щось не так”. Зазвичай, це відбувається одразу після народження, коли стають помітні аномалії очей. Більшість лікарів не знають, що таке синдром Лоу, отож багато сімей, особливо тих, де не було попередніх випадків захворювання, проходять довгий шлях, доки їм виставляється правильний діагноз та призначається лікування.
Коли ж вони нарешті дізнаються про діагноз сина, на них чекає шок, можливо вони не одразу повірять, що таке можливо. Тому слід одразу сказати їм, що ця хвороба ще не дуже досліджена і не можна сказати напевно нічого ані про можливий розвиток дитини, ані про тривалість її життя.
Після того, як улагоджені медичні проблеми і синові починають надавати відповідну допомогу, батькам слід звернути свою увагу на освіту та виховання їх дитини. Дитина, що потребує так багато уваги, зазвичай вимагає більше часу, енергії та грошей.
Такі родини з часом можуть стати соціально ізольованими та емоційно нестабільними, зосереджуючи усю свою увагу на хворій дитині. Хлопчики із синдромом Лоу спокійні та веселі, однак часом вони можуть бути роздратованими. Вони можуть влаштувати істерику з будь-якої дрібниці. Вони дуже цікаві співрозмовники. Періоди веселості в них можуть раптово змінюватися на напади впертості і небажання слухатись.
Лише невелика кількість батьків може остаточно прийняти факт того, що трапилося із їх дитиною. Протягом багатьох років вони переживають емоційні спади та підйоми. Однак, майже усі вони роблять необхідні зміни в житті для того, щоб приділяти більше уваги своїй дитині.
Ось декілька загальних порад батькам:
- Намагайтесь дізнатися якомога більше про синдром Лоу. Якщо у вас є інші діти, вони також мають знати все про хворобу їх брата.
- Намагайтеся розпочати лікування вашої дитини якомога раніше. Пам’ятайте, що це дуже тривалий і часом болісний процес.
- Прислухайтеся до порад лікаря та членів вашої родини. Не закривайтеся і залучайте усю свою сім’ю до нагляду за дитиною. Знову ж таки, якщо у вас є інші діти, ви можете відчувати свою провину за те, що приділяєте більше уваги хворому. Розмовляйте з ними, поясніть усю складність ситуації і нагадайте їм, що ви все рівно любите їх.
- Беріть участь у роботі батьківських груп та комітетів. Діліться із ними своїми знаннями та досвідом. Не ізолюйте себе.
- Знайте соціальні права своєї дитини та свої власні. Пам’ятайте, що насамперед ви самі є захисником вашої дитини.
- Не зважайте на свою підвищену емоційність – це нормально. Немає правильних чи неправильних почуттів. Важливо те, на що ви скеровуєте свої емоції.
Переглянуто редакційною колегією I.B.I.S.: 05/02/2002
Синдром Жильбера
(Gilbert Syndrome)
Тетяна Гаркун
Інформаційний спеціаліст Хмельницького ОМНІ-Центру
Коли вперше описали синдром Жильбера?
Августин Жильбер та П’єр Леребулей в 1901 році вперше описали захворювання, основною клінічною рисою якого є некон’югована гіпербілірубінемія, яке згодом на честь автора було названо синдромом Жильбера.
Які симптоми даного захворювання?
У принаймні 30% пацієнтів перебіг хвороби безсимптомний, хоча бувають неспецифічні симптоми, такі як спазми в животі, втомлюваність, слабкість. Часто вони супроводжуються відчуттям тривоги. Ці ознаки самі з часом зникають і потребують лише підтримуючого лікування.
Які причини виникнення синдрому Жильбера?
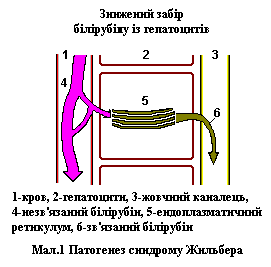
Однією з основних причин виникнення цього захворювання є підвищення рівня білірубіну, внаслідок зниженої активності фермента (білірубін-уридин-дифосфат-глюкороніл-трансфераза), що приймає участь у його перетворенні. В свою чергу зниження активності обумовлено наявністю мутації в гені, що кодує цей фермент. Цей фермент причетний також до метаболізму широкого кола субстратів (карциногени, ліки, гормони та нейротрансміттери). Тому вживання деяких медикаментозних препаратів, зміна гормонального фону може провокувати прояви синдрому Жильбера. Патогенез синдрому Жильбера дивіться на малюнку 1.
Як часто зустрічається синдром Жильбера?
Дані є неоднорідними. У США синдром Жильбера діагностують у 3-7% населення. Частіше захворювання зустрічається у африканців (36%), і лише у 3% мешканців Азії.
Який перебіг цієї хвороби?
Синдром Жильбера є відносно легким захворюванням, що не призводить до летальних випадків та значного погіршення стану здоров’я. Проте описані випадки гострих кризів, спровокованих медикаментозними препаратами, оперативним втручанням, голодуванням, тощо.
Чи з однаковою частотою хворіють чоловіки і жінки даним синдромом?
Синдром Жильбера трапляється переважно у чоловіків і співвідношення частоти захворювання чоловіків до жінок коливається від 2:1 до 7:1. Синдром Жильбера не обмежується якоюсь етнічною групою і властивий представникам усіх рас.
У якому віці найчастіше виявляють синдром Жильбера?
Синдром Жильбера найчастіше діагностується у неонатальному періоді та у підлітків в період статевого дозрівання. У людей старшого віку даний діагноз зазвичай встановлюється випадково при виявленні жовтушності (некон’югованої гіпербілірубінемії), що провокується випадковою хворобою або стресом.
Які фактори сприяють розвитку проявів синдрому Жильбера?
- Зневоднення.
- Голодування.
- Вірусна інфекція.
- Менструації.
- Стреси (фізичні та психічні навантаження).
- Вживання ряду медикаментів (гормональні контрацептиви, сульфаніламідні препарати).
Які аналізи необхідно провести для виявлення синдрому Жильбера?
- Загально-клінічний аналіз крові (включаючи ретикулоцити та білірубін).
- Визначення рівня лактатдегідрогенази.
- Печінкові тести.
Як лікується синдром Жильбера?
Найважливішим аспектом лікування, якщо діагноз встановлений, є розуміння того, що синдром Жильбера має відносно сприятливий перебіг і рідко супроводжується значним погіршенням якості та тривалості життя. Різні медикаменти використовуються експериментально для зниження рівня гіпербілірубінемії при синдромі Жильбера.
Чи госпіталізують хворих на синдром Жильбера?
Госпіталізація, здебільшого, не потрібна.
Чи потрібно дотримуватись певної дієти при даному синдромі?
Ні. Особливі застереження щодо запобігання тривалому голодуванню та при прийомі деяких ліків.
Чи потрібно хворим якісь обмеження в фізичній активності?
Ні. Не потрібні жодні обмеження активності.
Як запобігти цій хворобі?
Необхідно уникати відомих факторів, що прискорюють розвиток гіпербілірубінемії (наприклад, зневоднення, голодування). Якщо ви приймаєте якісь медикаменти, обов’язково повідомте вашого лікаря, що їх приписав, про ваш діагноз.
Яким є прогноз?
Щодо тривалості життя є сприятливим. Синдром Жильбера не наносить відчутної шкоди для хворих, і тому останні можуть проводити нормальне життя.
Переглянуто редакційною колегією I.B.I.S.: 28/10/2004
Дивіться також:
Синдром Жильбера
(Gilbert Syndrome)
Наталія Зимак-Закутня
Завідуюча Хмельницькою
медико-генетичною консультацією
Синоніми:
Вроджена гіпербілірубінемія, сімейна негемолітична жовтяниця, спадкова негемолітична білірубінемія.
Історія:
 Августин Жильбер та П’єр Леребулей в 1901 році вперше описали захворювання, основною клінічною рисою якого є некон’югована гіпербілірубінемія, яке згодом на честь автора було названо синдромом Жильбера. Жильбер описав характерну тріаду клінічних проявів: “печінкова маска” (жовтяниця), ксантелазми повік, періодичність симптомів з типовим посиленням жовтяниці після інфекцій, емоційного та фізичного перевантаження, після голодування.
Августин Жильбер та П’єр Леребулей в 1901 році вперше описали захворювання, основною клінічною рисою якого є некон’югована гіпербілірубінемія, яке згодом на честь автора було названо синдромом Жильбера. Жильбер описав характерну тріаду клінічних проявів: “печінкова маска” (жовтяниця), ксантелазми повік, періодичність симптомів з типовим посиленням жовтяниці після інфекцій, емоційного та фізичного перевантаження, після голодування.
Гортаючи сторінки літературної класики, око досвідченого лікаря [5,6] знаходить характерні ознаки синдрому Жильбера у лермонтовського Печоріна – інтермітуюча жовтяниця, яка провокувалась нервово-психічним збудженням і не впливала на загальний стан (“Я повернувся додому… отруйна злість мало-помалу заповнювала мою душу… Я не спав всю ніч. До ранку я був жовтий, як помаранч”). Крім того, Печорін – молодий чоловік, чиї походеньки не викликають сумнівів у міцному здоров’ї, і поява жовтяниці, судячи з наступних подій, зовсім не знижує його життєвого тонусу. Печоріна хвилювали диспептичні явища. Події повісті “Княжна Мері” проходять у місцевості, де “шумлять цілющі джерела” (Кисловодськ, П’ятигорськ); Печорін приймає “належну кількість склянок “Нарзану” ( “…в мене препаршивий шлунок”). Печоріну притаманна емоційна лабільність – нерівність поведінки, схильність до депресії, імпульсивність бажань та вчинків.
Критерії діагнозу:
- Періодична жовтяниця при відсутності гемолізу або хвороб печінки, котра може бути спровокована зневодненням, голодуванням, менструацією, стресами, такими як випадкова хвороба або сильні фізичні навантаження.
- Гіпербілірубінемія, при якій спостерігаються незначні зміни рівня білірубіну протягом дня чи сезону, так що іноді рівень білірубіну може бути нормальним у третини пацієнтів.
- Вірусна інфекція.
- Пацієнти можуть скаржитись на невизначений дискомфорт в області живота або на загальну слабкість без відомих на це причин. Ці симптоми самі з часом зникають і потребують лише підтримуючого лікування.
Своєчасна діагностика синдрому Жильбера дозволяє уникнути чисельних додаткових, часто необґрунтованих обстежень, прийому різноманітних препаратів, що не показані пацієнту, до невиправданої втрати коштів та часу.
Патофізіологія:
Причиною непрямої гіпербілірубінемії при синдромі Жильбера є знижена активність кон’югуючого фермента системи білірубін-уридин-дифосфат-глюкороніл-трансферази (білірубін-УГТ). Білірубін-УГТ перетворює білірубін в білірубін-моноглюкуроніди і диглюкуроніди у ендоплазматичному ретикулумі гепатоцитів. Водночас білірубін-УГТ є однією з найважливіших форм-УГТ, що здійснюють глюкуронізацію широкого кола субстратів (карциногени, ліки, гормони та нейротрансміттери) [7].
 Знання про ці ферменти значно розширились після опису УГТ у людини. Ген, що відповідає за білірубін-УГТ локалізується на 2 хромосомі і має складну будову: містить 5 екзонів, з яких з 2-5 екзони на 3′ кінці є сталими (мал.1) [4].
Знання про ці ферменти значно розширились після опису УГТ у людини. Ген, що відповідає за білірубін-УГТ локалізується на 2 хромосомі і має складну будову: містить 5 екзонів, з яких з 2-5 екзони на 3′ кінці є сталими (мал.1) [4].
Екзон 1 кодує унікальну послідовність кожної УГТ і відповідає за специфічність до субстрату. Однак, існують множинні екзони 1 (принаймі 13): екзони 1а і 1в кодують варіабельну ділянку для білірубіну УГТ*1 (також відомого як УГТ1А1) і УГТ*2, відповідно, де УГТ*1 відповідає за кон’югацію всього білірубіну, а УГТ*2 відіграє меншу роль. Експрессія УГТ*1 залежить від промотора у 5′ позиції відносно до кожного екзону 1, що містить ТАТАА послідовність. Таким чином, пошкодження глюкуронізації білірубіну може бути спричинене мутаціями в екзоні 1а, його промоторі, або у екзонах 2-5.
Справжній прорив в розумінні генетичної основи синдрому Жильбера відбувся в 1995 році, коли вдалося ідентифікувати мутації в ТАТАА ділянці промотора. В гомозиготному стані, зменшення глюкуронізації білірубіну спостерігається тоді, коли у жовчі білірубін-диглюкуронід переважає над білірубін-моноглюкуронідом. З того часу також були ідентифіковані додаткові мутації. Наприклад, окремі азіатські пацієнти з синдромом Жильбера без видимих клінічних ознак не мають мутацій на рівні промотора, але є гетерозиготними за міссенс-мутаціями (Gly71Arg, Tyr486Asp, Pro364Leu) в кодуючому регіоні. Такі особи також мають значно вищий рівень білірубіну, ніж особи з алелем дикого типу. Механізм зниження активності білірубін-УГТ остаточно не з’ясований. Інші фактори, такі як гострий гемоліз або порушення транспорту в гепатоцитах відіграють певну роль у клінічній експресії синдрому Жильбера. Наприклад, багато осіб гомозиготних до дефекту ТАТАА не мають некон’югованої гіпербілірубінемії, а у пацієнтів зі зменшеним рівнем білірубін-УГТ, що характерно для деяких грануломатозних хвороб печінки, не розвивається гіпербілірубінемія.
Частота:
- У США: Синдром Жильбера діагностують у 3-7% населення.
- У всьому світі: частота захворювання синдромом Жильбера надто варіабельна залежно від критеріїв, які використовуються при діагностуванні: кількість визначень білірубіну, метод аналізу, рівень білірубіну, та знання того, чи голодував пацієнт. Нові дані щодо поширеності синдрому Жильбера привносяться молекулярно-генетичними дослідженнями поліморфізмів в ділянці ТАТАА. Такі поліморфізми, приміром, спостерігаються у 36% африканців, але лише у 3% азіатів. Клінічний фенотип може бути не таким очевидним як визначений генотип через впливи навколишнього середовища, такі як глюкуронізація білірубіну, викликана алкоголем, що знижує рівень білірубіну.
- Серед сімейних функціональних гіпербілірубінемій синдром Жильбера займає 1 місце.
Тип успадкування: аутосомно-домінантний.
Перебіг хвороби:
Синдром Жильбера є відносно легким захворюванням, що не призводить до летальних випадків. Перебіг хвороби, зазвичай, не потребує жодних обмеженнь фізичної діяльності та дозволяє проводити звичайний спосіб життя та харчування. Госпіталізація, як правило, не потрібна. Однак, такий перебіг можливий за умови уникнення дії відомих провокуючих факторів.
Провокуючі фактори:
- Зневоднення.
- Голодування, що викликає підвищення рівня некон’югованого білірубіну у плазмі.
- Інтеркуррентне захворювання, таке як вірусна інфекція.
- Менструації.
- Стреси (фізичні та психічні навантаження).
- Медикаменти (пероральні контрацептиви, сульфаніламіди, гепарин, саліцилати, анаболічні стероїди, глюкокортикоїди, андрогени, рифампіцин, циметидін, левоміцетин, стрептоміцин, ампіцилін, кофеїн, етиніл-естрадіол, парацетамол, тобто ті препарати, в метаболізмі яких приймає участь УДФГТ).
Співвідношення статей:
Синдром Жильбера трапляється переважно у чоловіків і співвідношення частоти захворювання чоловіків до жінок коливається від 2:1 до 7:1. Синдром Жильбера не обмежується якоюсь етнічною групою і трапляється в усіх расах.
Вік маніфестації:
Синдром Жильбера найчастіше діагностується в неонатальному періоді та у періоді статевого дозрівання, можливо через затримку глюкуронізації білірубіну ендогенними стероїдними гормонами. У людей старшого віку даний діагноз, зазвичай, встановлюється випадково при виявленні некон’югованої гіпербілірубінемії, що провокується випадковою хворобою або стресом.
Клініка:
Принаймні 30% пацієнтів мають безсимптомний перебіг хвороби, хоча бувають неспецифічні симптоми, такі як спазми в ділянці живота, втомлюваність, слабкість. Часто вони супроводжуються відчуттям тривоги. Спостерігається підвищена реактивна та особиста тривожність, погане самопочуття та зниження активності, порушення біоритму, розвивається астенічний синдром. У деяких випадках при синдромі Жильбера виявляють помірне збільшення печінки, холецистит, у тому числі калькульозний, холангіт, дисфункцію жовчного міхура та сфінктера Одді (переважно по гіпомоторному типу).
Майже у всіх хворих виявляються зміни біохімічного складу жовчі, що характеризують літогенність жовчі: зниження холево-холестеринового коефіцієнту і холато-холестеринового індексу, підвищення індексу літогенності. Тому пацієнтів із синдромом Жильбера відносять у групу ризику розвитку холелітіазу.
Поліформізм симптоматики та відсутність чіткого зв’язку із рівнем білірубіну в плазмі ускладнюють діагностику. Немовлята із синдромом Жильбера можуть мати більшу схильність до неонатальної жовтяниці при годуванні груддю або поєднанні з іншими спадковими порушеннями метаболізму.
Диференційна діагностика:
Синдром Жильбера потребує грунтовної діагностики для встановлення причин некон’югованої гіпербілірубінемії та відмежування від ряду станів, як от [1]:
- Синдром Мейленграхта;
- Синдром Дабіна-Джонсона;
- Синдром Ротора;
- Синдром Люсі-Дріскола;
- Синдром Криглера-Найяра;
- Гемоліз;
- Гематома;
- Гострі та хронічні гепатити;
- Холецистит, дискінезія жовчовивідних шляхів;
- Первинна гіпербілірубінемія через неефективний еритропоез;
- Недостатність глюкуроніл-трансферази;
- Інфекціі;
- Кардіальні хвороби (наприклад, застійна серцева недостатність, штучні клапани серця);
- Проживання на великій висоті над рівнем моря;
- Медикаментозне лікування (наприклад, пробенецидом, рифампіцином та іншими антибіотиками);
- Тиреотоксикоз.
Деякі диференціально-діагностичні критерії представлені у наступній таблиці [1]:
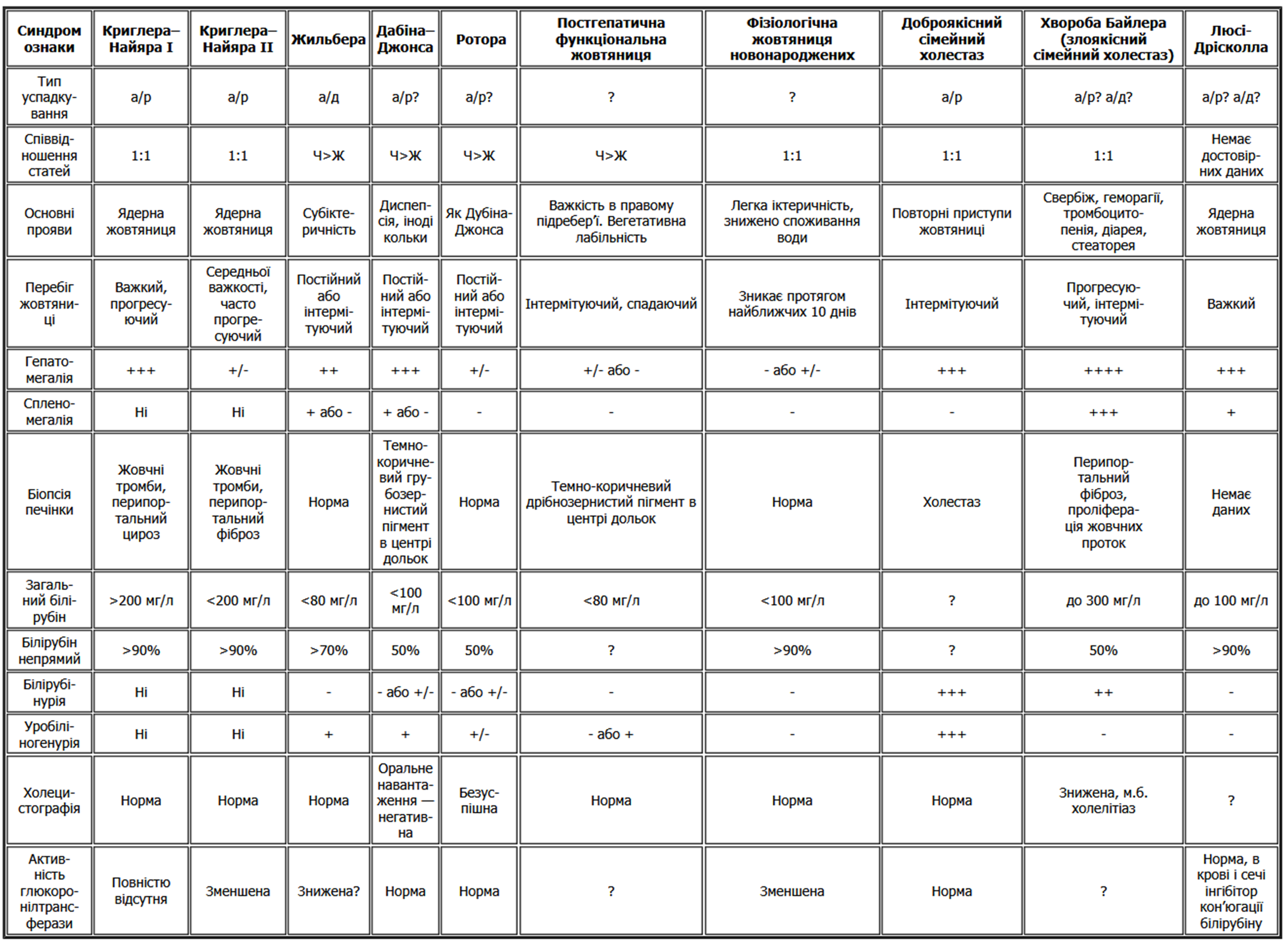
Патогенез синдрому Жильбера та деяких інших функціональних гіпербілірубінемій дивіться на малюнку 2 [2,4].

Лабораторні обстеження:
- Загально-клінічний аналіз крові, включаючи ретикулоцити.
- Білірубін та його фракції.
- Тести для виключення гемолізу.
- Визначення рівня лактатдегідрогенази, який залишається нормальним при синдромі Жильбера, проте підвищується при гемолізі.
- Печінкові тести: за вийнятком некон’югованої гіпербілірубінемії та підвищення рівня лужної фосфатази, патологічні зміни не характерні.
Інші аналізи:
Іноді поряд із новітніми молекулярно-генетичними технологіями використовують провокаційні тести, які, щоправда, представляють переважно історичний інтерес та мають низьку специфічність та достовірність.
Голодування. При голодуванні протягом 48 годин збільшується рівень некон’югованого білірубіну в плазмі у 2-3 рази та повертається до норми через 24 години після відновлення нормальної дієти. Хоча рівень некон’югованого білірубіну підвищується також при голодуванні пацієнтів з гемолізом або гепатитом, величина підвищення менша, ніж у хворих із синдромом Жильбера. Подібне підвищення білірубіну у плазмі також спостерігається при дієті з дефіцитом жирів та нормальним калоражем, однак рівень білірубіну нормалізується при відновленні вживання жирів. Точний взаємозв’язок голодування та гіпербілірубінемії, яка викликана дієтами, залишається неясним.
Нікотинова кислота. Внутрішньовенне введення 50 мг нікотинової кислоти спричиняє підвищення рівня некон’югованого білірубіну у плазмі в 2-3 рази через 3 години. Механізми виникнення цього є багатофакторними і, мабуть, стосуються: 1) зниженої резистентності червоних кров’яних тілець, 2) підвищення утворення білірубіну у селезінці, 3) транзиторного пригнічення активності білірубін-УГТ, та 4) підвищення активності гемоксигенази селезінки. Подібне, але менш виражене зростання рівня білірубіну може спостерігатись як у здорових людей, так і у пацієнтів із хронічними захворюваннями печінки або гемолізом, тому нікотиновий тест не є специфічним і не дозволяє чітко відмежовувати саме пацієнтів із синдромом Жильбера.
Фенобарбітал. Фенобарбітал та інші ензимні індуктори білірубінової-УГТ системи нормалізують білірубін у плазмі пацієнтів із синдромом Жильбера. Це переважно відбувається як через прискорений кліренс білірубіну плазми, так і через знижений метаболізм білірубіну. Стероїди можуть також зменшити рівень білірубіну у плазмі при синдромі Жильбера.
Тонкошарова хроматографія. Використовується для діагностики синдрому Жильбера, оскільки цей метод показує підвищене співвідношення білірубіну-моноглюкуроніду до білірубіну-диглюкороніду, відображаючи зменшену активність білірубін-УГТ, що дозволяє диференціювати синдром Жильбера від хронічного гемолізу, захворювань печінки.
Токсичні впливи. Оскільки детоксикація більшості ксенобіотиків відбувається шляхом глюкуронізації, дефект фермента білірубін-УГТ при синдромі Жильбера зумовлює ряд токсичних впливів при прийомі ряду медикаментів (пероральні контрацептиви, сульфаніламіди, гепарин, саліцілати, тощо).
Полімеразна ланцюгова реакція. Полімеразна ланцюгова реакція є швидким методом встановлення генетичних поліморфізмів в ТАТАА ділянці УГТ1*1 гена.
Біопсія печінки рідко є необхідною для встановлення діагнозу.
Гістологічні дані. Гістологічно печінка є нормальною. Зрідка спостерігається накопичення ліпофусциноподібного пігменту довкола термінальних печінкових венул.
Лікування:
Найважливішим аспектом лікування, якщо діагноз встановлений, є запевнення хворих про те, що синдром Жильбера має відносно сприятливий перебіг і рідко супроводжується значним погіршенням якості та тривалості життя.
Лікування проводиться за наступними принципами:
- Виведення кон’югованого білірубіну (посилений діурез, активоване вугілля як абсорбент білірубіну в кишечнику).
- Руйнування білірубіну, фіксованого в тканинах, тим самим вивільняються периферичні рецептори, які можуть зв’язувати нові порції білірубіну, попереджується його проникнення через гемато-енцефалічний бар’єр. Досягається це методом фототерапії. Максимальний ефект спостерігається при довжині хвиль до 450 нм. Лампи з синім світлом ефективніші, однак вони ускладнюють спостереження за шкірою дитини. Фото-джерело розміщують на відстані 40-45 см над тілом (процедуру проводити тільки в кювезі при контролі температури). Очі дитини необхідно захистити. Фотодеградацію білірубіну посилює рибофлавін, який є навіть у внутрішньоклітинній концентрації хромофором. Необхідна тривалість фототерапії зменшується і при додаванні холестираміну, хоча цей препарат менш фізіологічний, ніж рибофлавін. Фототерапія значно ефективніша при одночасному проведенні сеансів оксигенобаротерапії, оскільки кисень посилює декон’югацію білірубіну.
- Перспективним є застосування цитохромів.
- Запобігання дії провокуючих факторів (інфекцій, перенавантаженнь), препаратів- конкурентів глюкуронізації або тих, які витісняють білірубін із зв’язку з альбуміном (пероральні контрацептиви, сульфаніламіди, гепарин, саліцілати).
- Різні медикаменти використовуються експериментально для зниження рівня гіпербілірубінемії при синдромі Жильбера. Наприклад, фенобарбітал та глютатимід підвищують активність білірубін-УГТ, в той час, як тин-протопорфирін пригнічує гемоксигеназу, зменшуючи рівень білірубіну.
Прогноз:
Щодо тривалості життя є сприятливим. Синдром Жильбера не наносить відчутної шкоди для хворих, і тому останні можуть проводити нормальне життя.
Освіта пацієнтів:
Пацієнти повинні усвідомлювати, що жовтяниця може прогресувати, якщо вони будуть пропускати прийоми їжі, допускати зневоднення організму, при розвитку інфекцій (вірусних, наприклад), стресах.
Номер з каталогу МІМ:
143500 Gilbert Syndrome.
Література:
- Делягин В.М., Бурков С.Г. Семейные формы функциональных гипербилирубинемий в работе практикующего врача //Лечащий Врач.- 1998.- № 2.- С. 17-22.
- Губергриц Н.Б. Функциональные гипербилирубинемии //Doctor.- 2004.- № 3.- С.19-22.
- Губергриц Н.Б. Рациональная терапия функциональных гипербилирубинемий //Здоровье Украины.- 2004.- № 89.- С. 17.
- Губергриц Н.Б. Функциональные гипербилирубинемии: патогенез, клиника, диагностика, лечение //Здоровье Украины.- 2004.- № 89.- С. 15-16.
- Еселев М.М. Сцепуро П.Г. Синдром Жильбера.- Саратов: Изд-во Сарат.ун-та, 1991.- 70 с.
- Еселев М.М. Сцепуро П.Г. Чем болел Печорин? //Наука и жизнь.- 1992.- № 1.- С. 92-94.
- Mukherjee S. Gilbert syndrome (www.emedicine.com).
- Aono S, Adachi Y, Uyama E, Yamada Y, Keino H, Nanno T, Koiwai O, Sato H. Analysis of genes for bilirubin UDP-glucuronosyltransferase in Gilbert’s syndrome. Lancet. 1995 Apr 15;345(8955):958-9. [Medline]
- Bancroft JD, Kreamer B, Gourley GR. Gilbert syndrome accelerates development of neonatal jaundice. J Pediatr. 1998 Apr;132(4):656-60. [Medline]
- Burchell B, Hume R. Molecular genetic basis of Gilbert’s syndrome. J Gastroenterol Hepatol. 1999 Oct;14(10):960-6. [Medline]
Переглянуто редакційною колегією I.B.I.S.: 28/10/2004
Дивіться також:
Епігнатус
(Epignathus)
Світлана Бузаш
Лікар-гінеколог
Рівненської жіночої консультації № 1
Визначення:
Епігнатус – це тератома, яка розвивається з порожнини рота та глотки.
Патологічна анатомія:
Найчастіше пухлини розвиваються з клиноподібної кістки, деякі з твердого піднебіння, м’якого піднебіння, глотки, язика та щелепи. Розвиваючись, вони проростають в порожнину рота, носа або черепа. У більшості випадків пухлини доброякісні. За гістологічною будовою вони представляють утвори тканин одного або трьох зародкових шарів. Найчастіше вони складаються з жирової, хрящової, кісткової та нервової тканин. Пухлини можуть заповнювати порожнину рота та верхніх дихальних шляхів та призводити до гострої асфіксії безпосередньо після народження. В результаті обструкції порожнини рота плода вагітність може ускладнюватись багатовіддям.
Пренатальна діагностика:
Великих розмірів пухлина, яка виходить з порожнини рота, може вказувати на цю патологію. Можуть візуалізуватись кістковий компонент пухлини та ділянки кальцинації. Захворювання зазвичай супроводжується багатовіддям. Важливо ретельно вивчити анатомію головного мозку, оскільки пухлина може проростати в порожнину черепа.
Диференційна діагностика:
Диференційна діагностика проводиться з тератомами шийної ділянки, цефалоцеле та іншими пухлинами обличчя.
Асоційовані аномалії:
В 6% випадків зустрічаються поєднані аномалії. До них відносяться: розщілина губи, множинні гемангіоми обличчя, бранхіогенні кисти, гіпертелоризм, пупкова кила, вроджені вади серця. Аномалії обличчя швидше за все зумовлені механічною дією пухлини на структури обличчя, які розвиваються.
Частота:
В 2% випадків у дітей тератоми зустрічаються в носоглоточній ділянці (включаючи ділянку рота, мигдалики та основу черепа). Більшість з них виявлено у новонароджених.
Прогноз:
Багатовіддя – несприятлива діагностична ознака. Основною причиною смерті новонароджених буває асфіксія в результаті обструкції дихальних шляхів. Відомі випадки оперативного лікування зі сприятливим результатом. Не має даних про малігнізацію пухлини. Кінцевий результат залежить від розмірів утвору та ступеня задіяності в процес життєво важливих структур. Пухлини, які діагностовано в антенатальному періоді, можуть бути дуже великими.
Акушерська тактика:
Ведення вагітності в ІІІ триместрі залежить від аномалії. При великих розмірах пухлини у плода оптимальним методом родорозрішення вважається кесарський розтин. Для можливої інтубації новонародженого повинна бути присутня бригада досвідчених фахівців.
Література:
- Пренатальная диагностика врожденных пороков развития плода /Р. Ромеро, Дж. Пилу, Ф. Дженти, А. Гидини, Дж.С. Хоббинс.- М.: Медицина,1994.- C. 111-113.
- Gorlin RJ, Cohen MM, Hennekam RCM. Syndromes of the Head and Neck. Oxford University Press. 2001:970-971.
- Warkany J. Congenital Malformations. Notes and Comments. Year Book Publisher. Chicago. 1981:1241-1242.
Переглянуто редакційною колегією I.B.I.S.: 27/08/2003
|
|
|
|
|
|





