
Serhiy
Гіпофосфатемічний рахіт
(Hypophosphatemic Rickets)
Олег Степанович Зимак
Доцент кафедри педіатрії факультету удосконалення лікарів
Вінницького національного медичного університету ім. М.І. Пирогова
Синоніми і ключові слова:
Спадковий гіпофосфатемічний рахіт; рахіт, стійкий до вітаміну D, гіпофосфатемічний рахіт, зчеплений з Х-хромосомою; гіпофосфатемічна остеомаляція, зчеплена з Х-хромосомою.
Основні відомості:
Термін “рахіт” походить від давньоанглійського слова “wrick”, тобто розтягнення (викривлення). Це викривлення або деформація кісток відомі лікарям протягом багатьох століть. Поступово з’ясувалось, що воно є проявом ряду захворювань. З часу застосування на початку ХХ-го сторіччя вітаміну D та ультрафіолетового опромінення як ефективних профілактичних та лікувальних засобів при рахіті, з’ясувалось, що деякі форми рахітичних захворювань залишались резистентними (стійкими) до такого лікування. Вивчення подібних випадків виявило спільний фактор – низьку концентрацію фосфатів у сироватці. Більше того, стан цей мав схильність до спадкової передачі. Лікування таких пацієнтів вітаміном D навіть у достатньо високих дозах не приводило до одужання, звідси і виник термін “рахіт, стійкий до вітаміну D”. У 1958 році спадковий гіпофосфатемічний рахіт отримав формальне визначення “Х-зчеплений фосфатемічний рахіт”. Детальне та грунтовне дослідження виявило, що зниження рівня фосфату в сироватці – гіпофосфатемія – є неодмінним симптомом хвороби, проте у жінок кісткові ураження спостерігались рідше та мали м’якший перебіг, що, згідно з гіпотезою Лайон, може бути викликане вибірковою інактивацією Х-хромосом у жінок. Клінічна реакція на різні аналоги холекальциферолу дає підстави стверджувати, що дефектним фактором може бути порушення 1-альфа-гідроксилування 25-гідроксихолекальциферолового метаболіту, що виробляється у печінці.
Патогенез:
За останні роки було досягнуто значних успіхів, зокрема у випадку клонування мутантного гену під назвою PHEX. Вважається, що цей ген, локалізований на Х-хромосомі (Xp22.2–p22.1), кодує поки невідомий гормон, що регулює обмін фосфатів.
Підвищена втрата фосфатів на рівні проксимальних ниркових канальців приводить до порушення процесів нормальної осифікації. Цей феномен є вторинним відносно порушення функції фосфатно-натрієвого каналу “щіткової кайми” епітелію кишківника. Недостатній рівень неорганічних фосфатів порушує функціонування остеобластів – тобто клітин, що зумовлюють осифікацію кісткового матриксу, оскільки для формування дорослої кістки необхідно відкладення кристалів гідроксиапатиту [3Ca3(PO4)2 : Ca(OH)2]. Проте всі спроби застосування у комплексі лікування фосфатів зазнали невдачі.
Вживання фосфатів зумовлює посилене використання кальцію у процесах мінералізації кісток, а відтак зниження його рівня у сироватці крові, на що негайно реагує гормон паращитовидних залоз (паратгормон). Однак відновлення порушеного status quo швидко відбувається за рахунок пригнічення реабсорбції фосфатів нирковими канальцями. Тривале вивчення цього захворювання, разом із вражаючими успіхами, залишає ще багато “білих плям”.
Частота: невідома.
Стать:
- Хоча рівень фосфату в сироватці однаково понижений як у чоловіків, так і у жінок, у гетерозиготних жінок ступінь вираженості кісткових змін значно менший.
- Всі гемізиготні чоловіки є клінічно хворими.
Вік прояву:
- Як і при всіх генетичних розладах, хвороба присутня з моменту зачаття.
- Вага новонародженого загалом є нормальною, але у ранньому віці може спостерігатись сповільнення ростових показників. При народженні, зазвичай, відзначають черепний синостоз та підвищену щільність кісток.
Клініка та діагностика:
- Першою клінічною ознакою гіпофосфатемічного рахіту є дещо уповільнений темп росту протягом першого року життя.
- Нездатність пацієнта підтримувати вагу тіла при перших спробах стояти чи ходити.
- Пацієнти не мають судом чи інших системних ознак м’язової дисфункції або порушень окислювального метаболізму.
- Зважаючи на те, що є хворі гетерозиготні жінки, можливо, що серед родичів по материнській лінії зустрічаються випадки низького зросту та рахіту. У чоловіків також очікується низький зріст.
- Характерне пізнє прорізування зубів, множинні абсцеси.
- Інтелектуальний розвиток не порушений.
- Викривлення довгих кісток нижніх кінцівок (що несуть вагу) стає очевидним наприкінці першого–другого року життя.
Диференційна діагностика проводиться із такими захворюваннями, як:
- Цистиноз (OMIM 219800, 219900).
- Синдром Фанконі (OMIM 134600).
- Нирковий канальцевий ацидоз (OMIM 179830, 267200, 602722).
- Спадковий гіпофосфатемічний рахіт з гіперкаільціурією (OMIM 241530).
- Вітамін D–залежний рахіт (І і ІІ типи) – OMIM 277440.
- Псевдогіпопаратиреоз (OMIM 103580, 203330).
- Гіпофосфатемічний рахіт аутосомно-рецесивний (OMIM 241520).
- Гіпофосфатемічний рахіт аутосомно-домінантний (OMIM 193100).
Лабораторні дослідження:
- Починати лабораторну діагностику рахіту слід з оцінки рівнів сироваткового кальцію, фосфору та лужної фосфатази.
- При гіпофосфатемічному рахіті рівень кальцію може бути в межах чи дещо нижчим відповідних вікових норм; рівень лужної фосфатази значно перевищує норму.
- Ретельної та зваженої оцінки вимагає рівень фосфатів сироватки крові у дітей першого року, оскільки їх концентрації у немовлят (5,0-7,5 мг/дл) є високими порівняно з дорослими (2,7-4,5 мг/дл), тому дуже легко не помітити гіпофосфатемію.
- Рівень паратгормону знаходиться в межах відповідних норм чи трохи вищий, в той час як рівень кальцитріолу низький.
- Важливо: підвищена втрата фосфатів із сечею!
- Для контролю лікування важливо періодично проводити ультразвукові дослідження з метою вчасного виявлення розвитку нефрокальцинозу (ниркового кальцинозу). Зазвичай вважаючись наслідком хвороби, зараз це ускладнення визнано secuela лікування.
- Спостереження за співвідношенням кальцію до креатиніну. Співвідношення більше ніж 0,25 до 1 вимагає зменшення дози вітаміну D з метою запобігання нефрокальцинозу.
- Нирково-канальцева реабсорбція фосфатів – це співвідношення фосфатного кліренсу (ФК) до кліренсу креатиніну (КК).
- Фосфатний кліренс (ФК) розраховується за формулою:
фосфати в плазмі (мг/дл)
Замінивши фосфатні значення креатиніновими за цією ж формулою можна розраховувати кліренс креатиніну.
- Для розрахунків можна використовувати тільки ранкову сечу, оскільки в результаті ділення ФК на КК одиниці об’єму сечі взаємно скорочуються.
- Нирково-канальцева реабсорбція фосфатів при Х-зчепленій гіпофосфатемії складає 60%; нормальний же її рівень перевищує 90%.
Лікування:
Звичайні препарати вітаміну D для лікування цього розладу не підходять, оскільки їм бракує важливої активності 1-альфа-гідроксилази. Використання високих доз вітаміну D (в межах 25000-50000 МО/день – на нижній межі токсичної дози) спричиняло виникнення у пацієнтів частих гіперкальцемічних епізодів. Сьогодні стандартним призначенням для лікування спадкового рахіту є використання 1,25-дигідрокси-вітаміну D – кальцитріолу (синоніми – Рокалтрол, Кальцекс), що дозволяє уникнути токсичної дози останнього, запобігає значному накопиченню вітаміну D і зменшує небезпеку гіперкальцемії (симптомами гіперкальцемії є слабкість, нудота, біль у м’язах, запор. Кальцитріол збільшує рівень кальцію, сприяючи абсорбції кальцію в кишківнику та затримці у нирках. Доросла та дитяча доза становить 50 мг/кг/день, доза не змінюється щонайменше протягом 4 тижнів, можна збільшувати дозу на 5 мг/кг/день; але не більше 65-70 мг/кг/день.
Масивна втрата фосфатів із сечею є невід’ємною проблемою цього розладу, отже їх потрібно відновити за допомогою замінників фосфатів:
- Фосфатні солі (Neutra – Phos – K) – розчин замінника нейтралізованого зв’язаного фосфату, що містить 1000 мг (фосфору) Р (32 ммоль неорганічного фосфату) на 300 мл або 4 капсули чи пакети. Комбінація фосфатів Na і K. Доросла доза препарату становить 1-3 г/день 1% фосфору, тобто, 4-12 капсул чи пакетів/день, розчинивши вміст кожної капсули чи пакету у 75 мл води. Дитяча доза – 10 мг/кг/день фосфату; збільшення дози слід продовжувати до отримання концентрації фосфатів в сироватці >4,5 мг/дл (немовлятам); 2 мг/дл (дітям), розчинивши вміст капсули чи пакету у 75 мл води. Варто пам`ятати, що магній та алюміній-вмісні антрацити можуть зв’язувати фосфати і зменшувати рівні фосфатів у сироватці.
Протидіють тенденції втрати кальцію в кістках, тобто мають антикальційуричний ефект, діуретики-тіазиди:
- Дихлоротіазид (Esidrix, HydroDIURIL) – відомий діуретик з протигіпертонічною дією, перешкоджає реабсорбції натрію в дистальних канальцях, спричиняє підвищене виділення натрію і води, а також іонів калію і водню. Не засвоюється і швидко виводиться з сечею. Доросла доза препарату становить 25 мг/день внутрішньо; не більше 100 мг/день. Дітям віком до 6 місяців призначають по 3 мг/кг/день, старше 6 місяців до 2 років 1-2 мг/кг/день, але не більше 30 мг/день, старше 2 років – 1-2 мг/кг/день, але не більше 100 мг/день. Застереження при ниркових захворюваннях; хворобах печінки, подагрі, цукровому діабеті, та еритематозі; виснаження електролітів є природнім результатом застосування тіазиду, якому можна запобігти за допомогою ретельного спостереження за електролітами сироватки, особливо в жарку погоду; основною побічною дією є гіпокаліемія, тому бажаним є вживання домішків калію.
Амілорид і гідрохлортіазид призначаються для збільшення реабсорбції кальцію і для зменшення ризику нефрокальцинозу.
Хірургічна допомога:
- Остеотомія для виправлення кісткових деформацій нижніх кінцівок може бути необхідною дітям із пізно вставленим діагнозом, або якщо початкове лікування виявилось неадекватним.
- Спонтанні абсцеси часто вимагають стоматологічних процедур.
- Хворим особам слід уникати занять контактними видами спорту до повного вилікування рахіту.
Нагляд:
Регрес рахітичних змін зазвичай відбувається через 6-8 тижнів від початку призначеного лікування. Щотижневого контролю вимагає рівень кальцію і фосфатів у сироватці впродовж перших 2-3 місяців лікування. Також важливо спостерігати за виділенням кальцію і фосфатів із сечею.
Ускладнення:
- Характерна риса спадкового гіпофосфатемічного рахіту – низький зріст -диспропорційний внаслідок деформації та вкорочення нижніх кінцівок. Повідомляється про застосування гормону росту для ліквідації вказаних змін. Проте значна вартість курсу лікування та короткочасний його ефект суттєво обмежують широке застосування цього методу.
- Гостра гіперкальцемія (та її прояви – роздратованість, збудження і судомні напади) може виникнути під час лікування.
- Нефрокальциноз, як результат тривалої агресивної терапії, спостерігається у 47% пацієнтів, проте не прогресує до ниркової недостатності.
Прогноз:
Прогноз щодо життя та стану здоров’я при правильному лікуванні сприятливий.
Медико-генетичне консультування:
- Покликане допомогти батькам хворої дитини зрозуміти спадкову природу цього стану, методи діагностики та профілактики.
- Пацієнт і сім’я повинні розуміти важливість подальшого спостереження для уникнення ускладнень.
Лінки:
Номер з каталогу МІМ:
307800 Hypophosphatemic Rickets, X-Linked Dominant.
Література:
- Беникова Е.А., Бужиевская Т.И., Сильванская Е.М. Генетика эндокринных заболеваний. – К: Наукова думка, 1993. – С. 89-90.
- Медицина дитинства /Г.Р. Акопян, Ю.Г. Антипкин, В.І. Березінь та ін.- За ред. П.С. Мощича.- Навч. посібник: ун-т. – К: Здоров’я, 1994. – Т. 1. – С. 605–608.
- Baroncelli GI, Federico G, Bertelloni S, et al: Assessment of bone quality by quantitative ultrasound of proximal phalanges of the hand and fracture rate in children and adolescents with bone and mineral disorders. Pediatr Res 2003 Jul; 54(1): 125-36.
- Blackard WC, Robinson RR, White JE: Familial hypophosphatemia: report of a case, with observations regarding pathogenesis. New Engl J Med 1962; 266: 899-905.
- Chesney RW, Mazess RB, Rose P: Long-term influence of calcitriol (1,25-dihydroxyvitamin D) and supplemental phosphate in X-linked hypophosphatemic rickets. Pediatrics 1983 Apr; 71(4): 559-67.
- Dixon PH, Christie PT, Wooding C: Mutational analysis of PHEX gene in X-linked hypophosphatemia. J Clin Endocrinol Metab 1998 Oct; 83(10): 3615-23.
- Glorieux FH, Scriver CR, Reade TM: Use of phosphate and vitamin D to prevent dwarfism and rickets in X- linked hypophosphatemia. N Engl J Med 1972 Sep 7; 287(10): 481-7.
- Jones, Kenneth, Lyons: Smith’s recognizable patterns of human malformation – 5 th ed. 1997; 680.
- Kainer G, Chan JC: Hypocalcemic and hypercalcemic disorders in children. Curr Probl Pediatr 1989 Oct; 19(10): 489-545.
- Petersen DJ, Boniface AM, Schranck FW: X-linked hypophosphatemic rickets: a study (with literature review) of linear growth response to calcitriol and phosphate therapy. J Bone Miner Res 1992 Jun; 7(6): 583-97.
- Stickler GB, Morgenstern BZ: Hypophosphataemic rickets: final height and clinical symptoms in adults. Lancet 1989 Oct 14; 2(8668): 902-5.
- Winters RW, Graham JB, Williams TF, McFalls VW, Burnett CH: A genetic study of familial hypophosphatemia and vitamin D-resistant rickets with a review of the literature. Medicine 1958; 37: 97-142.
Переглянуто редакційною колегією I.B.I.S.: 24/05/2005
Ріст плоду
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”.
(тижні) |
(гр) |
(см) |
(см) |
(см) |
(см) |
|---|---|---|---|---|---|
Числа в дужках означають 1 стандартне відхилення (1 SD).
1Вимірювання дітей білої раси, народжених на рівні моря за Usher та McLean (J Pediatr 74: 901, 1969).
2Середнє значення тім’ячково-куприкової довжини вирахуване з довжини тіла за методом Scammon та Calkins (The Development and Growth of the External Dimensions of the Human Body in the Fetal Period, University of Minnesota, 1929).
3Довжина ступні здорової єврейської дитини, народженої в Ізраілі за Merlob та співавт. (Am J Dis Child 138: 140, 1984). Середні значення екстрапольовані для 26 та 42 тижнів.
Ріст та розвиток
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”.
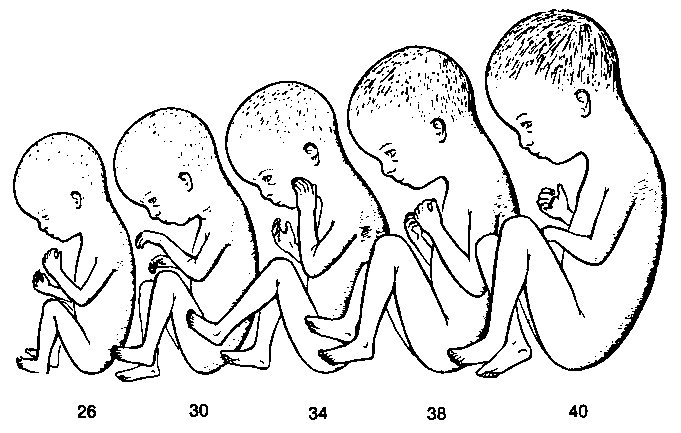
(тижні) |
(см) |
|
|---|---|---|
Відносні розміри плода у терміні 26-40 тижнів (згідно дати останньої менструації) та морфологічні характеристики, що допомагають встановити вік плоду. Дані взяті з книги Moore (The Developing Human, ed. 4, W.B.Saunders Co., 1988).
М’язова гіпотонія в дитинстві: гнучке дитя
(Muscle Hypotonia in Childhood)
Л.С. Євтушок
Зав. медико-генетичною консультацією
Рівненського клінічного лікувально-діагностичного центру
Визначення:
Гіпотонія визначається як відсутність м’язового опору при розтягуванні.
Етіологія:
Гнучкість у дитини може мати різну етіологію. Причиною її можуть бути ураження нервової системи на різних рівнях, починаючи від головного мозку і закінчуючи спинним мозком, невропатії, нервово-м’язові порушення та міопатій. Виділяють дві основні категорії причин (таб.1):
- паралітична гіпотонія (ураження периферичної нервової системи або нижчих моторних нейронів);
- непаралітична гіпотонія (ураження центральної нервової системи або центральних моторних нейронів).
Таблиця 1.
Основні причини м’язової гіпотонії в дитинстві.
|
Захворювання центральної нервової системи
|
Захворювання периферичної нервової системи
|
|---|---|
|
|
Більшість форм неонатальної гіпотонії мають центральне походження: гіпоксично-ішемічна енцефалопатія, внутрішньочерепні крововиливи, інфекції, гіпоглікемія, гіпотиреоз, хромосомні аномалії та вроджені порушення метаболізму.
Підходи до діагностики:
Зважаючи на те, що гіпотонія є дуже загальним і неспецифічним симптомом при захворюваннях новонароджених дітей, її діагностика вимагає дуже уважного обстеження, включаючи аналіз родоводу, перебіг вагітності та анамнез хвороби, об’єктивний огляд, лабораторні та інші діагностичні процедури.
При визначенні етіології дитячої гіпотонії насамперед необхідно встановити її характер – центральна чи периферична.
- Анамнез. Детально зібраний анамнез може допомогти у визначенні причини гіпотонії. Наприклад, вказівки в родоводі на випадки нервово-м’язових захворювань чи міопатії, знижену активність плода чи полігідрамніон під час вагітності, патологічні пологи (сідничне чи нижнє передлежання).
- Об’єктивний огляд. При огляді пацієнта необхідно проводити пошук характерних дизморфічних ознак, які можуть вказати на певний синдром та допомогти визначити ступінь і тип гіпотонії. Оцінка м’язового тонусу здійснюється за протидією при пасивних рухах суглобів. М’язовий тонус змінюється із віком. Недоношена дитина, наприклад, має відносну гіпотонію в порівнянні із доношеною. Гіпотонічні діти “падають крізь руки”, якщо утримувати їх під плечі. Якщо утримувати дитину, поклавши руку під грудну клітку, то дитина обвисне на руці оглядача. Дитині важко утримувати голову при сидінні. При об’єктивному огляді дитини необхідно встановити наявність вроджених контрактур; порушення смоктання чи ковтання, “пози жабки”, гепатоспленомегалії, ознак кардіоміопатичного синдрому.
- Гіпотонія центрального походження. Діти з центральною формою гіпотонії, як правило, мають інші порушення центральної нервової системи. Наприклад, відсутність реакції на зорові подразники, летаргія, затримка психомоторного розвитку, судоми або персистуючі рефлекси новонародженого (хапання, тонічний шийний тощо), підвищені сухожильні рефлекси чи міоклонус, спастика кінцівок. При типовому ураженні головного мозку діти погано контролюють рухи головою, мають слабкість у стегнах/плечах. При цьому спостерігається спастика в кінцівках та нормальні або підвищені сухожильні рефлекси. Дуже часто дитячому церебральному паралічу пацієнтів передує внутрішньоутробна або перинатальна травма (інсульт). При гіпотонії, що спричинена мозковим дизгенезом, в пацієнтів можуть спостерігатися дизморфічні риси. При таких відомих синдромах, як синдром Дауна і синдром Прадера-Віллі, гіпотонія вважається однією з основних ознак. Гіпотонія у новонароджених дітей домінує лише при декількох вроджених порушеннях метаболізму: молочно-кислий ацидоз, дефіцит піруват карбоксилази/дегідрогенези (порушення дихального ланцюжка), дефекти циклу сечовини, некетонна гіпоглікемія та пероксисомні хвороби. Крім гіпотонії у таких пацієнтів виникають інші симптоми: летаргія, кома, корчі, ацидоз тощо. Жодне вроджене порушення метаболізму в новонароджених не проявляється у вигляді ізольованої гіпотонії.
- Порушення периферичної нервової системи або функцій нижнього моторного нейрону (включаючи клітини передніх рогів периферичний нерв, нервово-м’язові синапси або м’язи) проявляється вираженою слабкістю з відсутніми або зниженими сухожильними рефлексами, м’язовою атрофією (яку важко діагностувати в дітей з надмірною вагою та ожирінням) інколи з фасцикуляцією язика. При об’єктивному огляді необхідно звертати увагу на інші симптоми, які можуть допомогти в класифікуванні гіпотонії. Так, слабкість м’язів обличчя є характерною для певних типів вроджених міопатій, таких як вроджена міотонічна дистрофія. Птоз та офтальмоплегія зустрічаються при вроджених міопатіях та важкій міастенії, тоді як фасцикуляція язика характерна для прогресуючої спінальної аміотрофії (хвороба Вердіга-Гофмана). Закріп, відсутність реакції зіниць, інші ознаки ураження черепних нервів виникають при набутому дитячому ботулізмі.
- Діагностичні тести:
|
При центральній гіпотонії
|
При периферичній гіпотонії
|
|
|
Лікування та медико-генетичне консультування:
Лікування та медико-генетична консультація будуть залежати від специфічного діагнозу. Опис деяких порушень, що часто асоціюються з дитячою гіпотонією:
Синдром Дауна. Частота синдрому Дауна серед живонароджених у всьому світі приблизно 1:700. Частота його у плода в три рази вища за цей показник (приблизно 2/3 вагітностей із синдромом Дауна закінчується спонтанним абортом). Клінічна діагностика захворювання базується на множинних малих аномалій і кількох великих. Ось деякі аномалії, що можуть зустрічатися в пацієнтів і бути пов’язаними із синдром Дауна: брахіцефалія, епікант, монголоїдний розріз очей, маленькі вушні раковини, плями Брушфільда, маленький ніс із запалим переніссям, маленький рот з висунутим язиком, коротка шия з надлишком шкіри, 4-х пальцева долонна борозна, клінодактилія п’ятих пальців з єдиною згинальною міжфаланговою складкою, що спричинено гіпоплазією середньої фаланги, широкий проміжок між 1-м та 2-м пальцями ноги. Кількість аномалій змінюється в кожному окремому випадку.
Іншими ознаками синдрому Дауна є сухість шкіри, відносно коротка статура, гіпотонія у дитинстві, передчасне старіння, широкі та пласкі крила клубових кісток та широкі кути вертлужної западини, що видно на рентгенограмах. Підлітки чоловічої статі мають знижений статевий потяг (лібідо) і, зазвичай, є стерильними, тоді як дівчата-підлітки можуть мати нормальне лібідо і спроможні вагітніти та народжувати. Приблизно третина дітей, народжених живими від таких матерів може мати синдром Дауна.
Основними великими аномаліями, що часто асоціюються із синдромом Дауна є вроджені вади серця (приблизно в 45% випадків). Зазвичай це атріовентрикулярний канал та вади вентрикулярних перегородок. Тому усі новонароджені з синдромом Дауна підлягають обов’язковому обстеженню кардіологом та ехокардіографії. Приблизно 7% пацієнтів мають шлунково-кишкові аномалії, з яких найчастіше зустрічається атрезія дванадцятипалої кишки. У дітей із СД спостерігається підвищена частота порушень щитовидної залози. Тому вони потребують щорічного обстеження щитовидної залози з визначенням рівня Т4 і TТГ в крові. Ризик виникнення гострої лейкемії підвищений в 10-15 разів. Новонароджені діти з СД можуть мати транзиторну лейкемоїдну реакцію. За рідкими виключеннями, пацієнти з синдромом Дауна відстають в розумовому розвитку. Ступінь відставання буває різним, від 20 до 80 IQ.
Причиною синдрому Дауна в 95% випадків є трісомія 21, як наслідок мейотичного нерозходження. Зайва хромосома 21 передається від матері в 95% випадків і лише в 5% від батька. Ризик анеуплоїдії у дитини збільшуєтся із віком матері. Тому ризик для подружжя мати дитину з синдромом Дауна є прямо пропорційним віку матері (таб. 2).
Таблиця 2.
Ризик народження дитини з синдромом Дауна
|
Вік матері
|
Ризик
|
|
менше, ніж 25 років
25-39 30-34 35-39 40-42 більше, ніж 42 |
1:1600
1:1100 1:700 1:250 1:80 1:40 |
Приблизно 25% повторних випадків народження в сім’ї дитини із трісомією 21 зумовлені мозаїцизмом – невеликим клоном клітин з трісомією 21 – в статевих клітинах одного з батьків.
Взагалі біля 1% випадків синдрому Дауна є наслідками хромосомного мозаїцизму.
Не завжди можна визначити механізм порушення в тому чи іншому випадку, однак ризик повторних випадків залежить від нього. Ризик повторного народження дитини з простою трісомією 21 складає 1% і має той самий механізм, що і перший випадок.
Приблизно 4% випадків синдрому Дауна представлені транслокацією між хромосомою 21 та іншою хромосомою. Може бути розрив хромосоми 21 з подальшим прикріпленням її довгого плеча іншої хромосоми (зазвичай, 14, рідко 13 або 15, іноді 22 або іншої 21). Такі аномалії можуть спостерігатися у сімейних випадках, коли один з батьків має збалансовану транслокацію (приблизно 1/3 випадків), тоді як усі інші випадки транслокації є спорадичними (de novo). Тому визначення каріотипу батьків є необхідним для з’ясування ризику народження дитини з трісомією 21.
Дуже рідко пацієнти із клінічними проявами синдрому Дауна мають нормальні хромосоми. Більшість таких випадків являють собою криптичну (субмікроскопічну) перебудову хромосом. Вона проявляється у трьох копіях ДНК, які розташовані в зоні 21q22.2-22.3 і мають назву критичної зони синдрому Дауна. Триплікація 50-100 генів в цій зоні спричиняє багато проявів синдрому Дауна (наприклад, гіпотонію, риси обличчя, серцеві вади). Таке криптичне порушення може бути виявлене при FISH-технології з використанням спеціальної проби. Подібне криптичне ураження може бути спричиненим як субмікроскопічною новою, так і успадкованою транслокацією, або нерівномірним кросинговером під час мейозу з залученням маленької зони довгого плеча хромосоми 21.
З метою діагностики синдрому Дауна існує можливість проводити пренатальні дослідження: амніоцентез і цитогенетичний аналіз культивованих амніоцитів на 16-18 тижні гестації, біопсію ворсин хоріона на 9-12 тижні гестації. Пренатальний скрінінг сироваткових маркерів крові підвищує можливості діагностики вагітностей з ураженим плодом. Комбіноване визначення рівнів альфа-протеїну некон’югованого естріолу та людського хоріонічного гонадотропіну дозволяє виявити 60% вагітностей плодом з синдромом Дауна.
Оскільки деякі прояви синдрому Дауна можуть загрожувати життю пацієнта, своєчасне медичне втручання підвищує тривалість їх життя. 50% пацієнтів з даною хворобою доживають до 50-річного віку. Медична допомога має бути скерована на подолання таких проявів, як гіпотиреоз та вроджені вади серця. В 12-20% випадків спостерігається несимптоматична нестабільність шийних хребців. Симптоми даного порушення, що пов’язані з компресією центрального нервового стовбура, є рідкісними. Література, присвячена радіографічному скринінгу, є дуже суперечливою. Хоча рентгенографія шийного відділу хребта може допомогти визначити нестабільність 1-2 шийних хребців, все ж неможна передбачити, хто з дітей буде мати проблеми із хребтом. Якщо в дитини з синдромом Дауна виникли порушення функції стравоходу чи сечового міхура, зміни в положенні шиї, необхідно провести неврологічне обстеження з обов’язковою рентгенографією шийного відділу хребта. У більшості пацієнтів такі симптоми проявляються у віці до 10 років, коли ослаблення зв’язок є найбільш важким.
Синдром Прадера-Віллі. Частота випадків синдрому Прадера-Віллі коливається від 1:10000 до 1:30000 серед живонароджених. Новонароджені з синдромом Прадера-Віллі зазвичай мають важку гіпотонію. Часто зустрічаються зниження рухової активності плода в утробі матері та сідничне передлежання. Порушення смоктання та ковтання можуть спричиняти задишку та викликати інші респіраторні проблеми. Характерний слабкий плач. Сухожильні рефлекси та рефлекс Моро зазвичай знижені. Відмічається затримка моторного розвитку. Більшість пацієнтів страждає на розумову відсталість різного ступеня (від легкого до важкого). Загальний рівень їх розумового розвитку коливається в межах від 35 до 85 IQ. Часто зустрічається затримка мовного розвитку. Гіпотонія зменшується протягом 2-3-го року життя, в пацієнтів розвивається неконтрольований апетит, який швидко призводить до ожиріння. Надлишок жиру відкладається в нижній частині тулуба, на сідницях та проксимальній частині кінцівок. Хоча обличчя не має характерних дизморфічних ознак, пацієнти мають коротку статуру, мигдалеподібні очі, зменшений біфронтальний діаметр, косоокість, гіпопігментацію (світле волосся, блакитні очі, дуже чутливу до сонячного світла шкіру у порівнянні з іншими членами родини), маленькі кисті рук та ступні. У чоловіків спостерігається гіпогонадизм. В жінок відмічається затримка або відсутність менархе; менструації є дуже нерегулярними. Рівень гонадотропіну зменшений, як у чоловіків, так і у жінок. Часто спостерігаються характерні плями на шкірі.
Синдром Прадера-Віллі є одним з синдромів порушення імпринтингу. При імпринтизі прояв функціонування гену залежить від кого з батьків він походить. Більшість пацієнтів (>70%) мають незначну інтерстиціальну делецію довгого плеча хромосоми 15 в зоні q11q13, яку можна діагностувати при хромосомному аналізі з високою розділяючою здатністю та FISH із використанням специфічних проб, що дозволяють візуалізувати регіон 15q11q13, який є послідовно порушеним в пацієнтів з синдромом Прадера-Віллі. В таких випадках делетованою є батьківська хромосома. Рідше випадки синдрому Прадера-Віллі (25%) пов’язані з материнською дисомією (дві материнських копії та жодної батьківської копії 15q). Для того, щоб встановити материнську дисомію необхідно провести спеціальні ДНК-тести.
В старшому дитячому віці при синдромом Прадера-Віллі проблема полягає в емоційній нестабільності хворих та несподіваних спалахах гніву та дратівливості. Аномальна та ненажерлива манера харчування є характерним проявом синдрому. Зменшена тривалість життя пояснюється кардіореспіраторними ускладненнями через важку форму ожиріння. Проблеми з поведінкою та переїданням часом можна виправити шляхом застосування інтенсивної програми корегування поведінки, що відбувається під батьківським контролем. Кількість калорій, що вживається має бути зменшеною за будь-яких умов. Чим важча ступінь ожиріння, тим частіше спостерігається діабетичний тип тесту толерантності до глюкози. Пацієнти з синдромом Прадера-Віллі частіше страждають на інсулинонезалежну форму цукрового діабету, яка піддається лікуванню оральними гіпоглікемічними препаратами.
Синдром Цельвегера. Синдром Цельвегера також відомий під назвою Церебро-гепато-ренальний синдром. Це одне з пероксисомних порушень. Пероксисомні хвороби являють собою групу порушень, в яких основною причиною патології є або порушення форми або недорозвиток пероксисоми, чи порушення функції одного з ензимів, що розташований в цій органелі. Синдром Цельвегера відноситься до 1-ї групи пероксисомних порушень.
В новонароджених дітей з синдромом Зельвегера крім до вираженої гіпотонії спостерігаються характерні дизморфічні риси, які легко розпізнати. Діагностичну цінність мають типові риси обличчя: високе чоло, монголоїдний розріз очей, гіпоплазія надбрівних дуг, епікант, велике тім’ячко, деформації зовнішнього вуха, гепатомегалія, аномалії очей (глаукома, катаракта або помутніння рогівки). Можуть спостерігатися ниркові кісти, серцеві аномалії (відкрита артеріальна протока, дефекти серцевої перетинки), корчі, церебральний дизгенез, контрактури кінцівок. Діагноз підтверджується аналізом жирних кислот з довгим ланцюгом, пипеколінової кислоти, фітанової та жовчних кислот в плазмі, та / або плазмогену в червоних кров’яних тільцях. Це аутосомно-рецесивне порушення з несприятливим прогнозом.
Міотонічна дистрофія (хвороба Стейнерта). Міотонічна дистрофія найчастіше проявляється в підлітковому або в дорослому віці. Часом вона зустрічається і у новонароджених та немовлят. Ця форма хвороби має характерні клінічні особливості, що відрізняють її від більш типової дорослої форми.
Основні клінічні ознаки вродженої міотонічної дистрофії:
- слабкість м’язів обличчя
- гіпотонія
- затримка моторного розвитку
- синдром дихальних розладів
- слабі рухи плода
- проблеми з годуванням
- клишоногість
- гідрамніон
- розумова відсталість
Типовою ознакою вродженої або дитячої міотонічної дистрофії є гіпотонія та диплегія обличчя. До інших ознак відносяться: слабкість щелеп, порушення дихання та годування в новонароджених. Розумова відсталість характерна для дітей старшого віку. Часто зустрічається стійка деформація стопи та артрогрипоз, що, як і інші прояви – м’язова дисфункція, гідрамніон, гіпоплазія діафрагми, тонкі ребра, зменшення активності плода – сформовані внутрішньоутробно.
В грудному та ранньому дитячому віці при вродженій міотонічній дистрофії ще відсутні не мають клінічних проявів міотонії. Тому можливе розходження в діагнозах щодо причини дитячої гіпотонії (спінальна м’язова атрезія, метаболічна міопатія, пероксисомні порушення тощо).
Диплегія обличчя та симптом “набитого” рота стають більш очевидними, коли дитина стає дорослішою. На другому десятку життя з’являються “дорослі” прояви хвороби.
Порушення дихання та підвищений ризик аспіраційної пневмонії часто призводять до ранньої смерті. Якщо дитина вижила протягом перших 3-4 тижнів, моторні функції можуть покращатись. Приблизно 2/3 пацієнтів, що вижили, будуть мати розумову відсталість. Довготривалий прогноз на майбутнє для таких пацієнтів не дуже сприятливий: неможливість самостійного життя, працевлаштування, підвищений відсоток смертності у віці до 30 років.
Молекулярний генетичний аналіз зараз є найкращим тестом для діагностики і може бути зроблений на основі проби крові. М’язова біопсія є корисною для клінічно нетипових випадків та випадків, коли може проявитись інше захворювання. Щодо електроміограми, то в ній немає потреби, коли аналіз ДНК дає чіткий результат. Часом, певних аналізів слід уникати при обстеженні дітей через їх болючість.
Генетичні аспекти міотонічної дистрофії. Ген міотонічної дистрофії знаходиться на хромосомі 19q. Мутаційна база цього порушення зумовлена нестабільністю послідовностей ДНК, що полягає в тринуклеотидних повторах. Міотонічна дистрофія з усіма її клінічними проявами (вроджена або доросла форма) також являє собою феномен антиципації та імпринтингу. Антиципація означає, що хвороба проявляється в більш ранньому віці та з більшим ступенем важкості в наступних поколіннях. Імпринтинг полягає в ефекті впливу на функцію гена батьківських похідних.
Тип успадкування міотонічної дистрофії – аутосомно-домінантний. Більшість вроджених випадків були зумовлені передачею від ураженої матері. На молекулярному рівні важкі форми вроджених випадків вказують на більшу кількість повторів. Клінічні ознаки міотонічної дистрофії не з’являються самі по собі як нові мутації, а успадковуються від одного з батьків, в якого є експансія повторів послыдовностей (CTG), але відсутні клінічні симптоми хвороби.
Пренатальна діагностика та визначення носія. Пренатальна діагностика найбільш необхідна для родин, в яких вже народжувалися діти із важкими проявами хвороби. Аналіз можна зробити протягом 10-12 тижня гестації за допомогою біопсії ворсин хоріону. Не варто давати точний прогноз, виходячи лише з результатів пренатального обстеження. Можливо, з часом можна буде більш точно вказати наскільки важким є ураження плода (менш вражений – “мінімальний фенотип” або важкий).
Щодо пацієнтів, які є клінічно здоровими, але мають експансію послідовних повторів хвороби, то прогноз для них не є однозначним. Кореляція між генотипом і фенотипом, зазвичай проявляється у ретроспективі. Тому прогноз треба давати дуже обережно. Мабуть, лише для премутаційних експансій (50-60 копій) можна впевнено сказати, що вони не спричинять симптомів нейром’язевих порушень. Для інших пацієнтів буде достатньо звичайного клінічного та офтальмологічного обстеження щорічно для підтримання нормального стану здоров’я.
Лікування. Пацієнти із міотонічною дистрофією можуть мати й інші аномалії (атрофію гонад, катаракту і порушення провідності серця). Медикаментозна терапія призначається лише для пацієнтів, в яких спостерігається важка ступінь міотонії (фенітоїн, прокаінамід чи хінін). Фенотоїн має тератогенні властивості, тоді як інші вказані ліки можуть спричинити порушення серцевої провідності.
Для більшості пацієнтів лікування зводиться до профілактики ускладнень та полегшення життя. Регулярний нагляд за порушеннями серцевої провідності, по можливості, уникання анестезії, застереження щодо аспіраційної пневмонії як результату ковтальної дисфункції, періодична контроль респіраторних функцій має велике значення.
Ураження клітин передніх рогів спинного мозку: синдром Вердніга-Гоффмана. Вроджена спінальна м’язова атрофія (синдром Вердніга-Гофмана) – це аутосомно-рецесивне захворювання, що спричинене дегенерацією клітин передніх рогів спинного мозку. Приблизно в третини пацієнтів хвороба проявляється вже в неонатальному періоді. При цьому в анамнезі є вказівки на зменшення рухливої активності плода внутрішньоутробно, наслідком цього часто є вроджені контрактури, вивих стегна. Ускладнення гіпотонії – загальна слабкість, відсутність рефлексів, знижений рефлекс смоктання та ковтання. Характерна “поза жабки”. В них завжди страдницький вираз обличчя.
Доброякісна вроджена гіпотонія. При доброякісній вродженій гіпотонії лабораторні аналізи не мають відхилення від норми. Гіпотонія присутня, але немає загальної слабкості та затримки розвитку. Розумовий розвиток також в межах норми. Рефлекси нормальні або дещо знижені. В деяких пацієнтів гіпотонія може зберігатися й у дорослому віці.
Література:
- Down syndrome: OMIM, #602523.
- Jones, K.L.: Recognizable Patterns of Human Malformation (5th ed.). W.B.Saunders Co, 771-772, 1997.
- Myotonic dystrophy: OMIM, #160900.
- Nyhan, W.L., Sakati, N.O.: Diagnostic Recognition of Genetic Disease. DNLM, 564-573, 1987.
- Prader-Willi syndrome: OMIM, #600162.
- Zellweger syndrome: OMIM, #214100.
- В підготовці даного інформаційного листка також використовувались лекційні матеріали курсу медичної генетики університету Південної Алабами.
Переглянуто редакційною колегією I.B.I.S.: 20/11/2002
Обвід голови у дівчат
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”.

Вимірювання обводу голови проводилось у дівчат віком від народження до 18 років. Загалом в рамках довготривалої програми спостереження Фелс протягом 1930-1982 років було обстежено 888 пацієнтів, які проживали в південно-західній частині штату Огайо. Усі пацієнти належали до різних соціально-економічних груп. Roche AF, Mukherjee D, Guo S et al: Head circumference reference data: Birth to 18 years, Pediatrics 79:706, 1987.
Обвід голови у хлопчиків
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”.
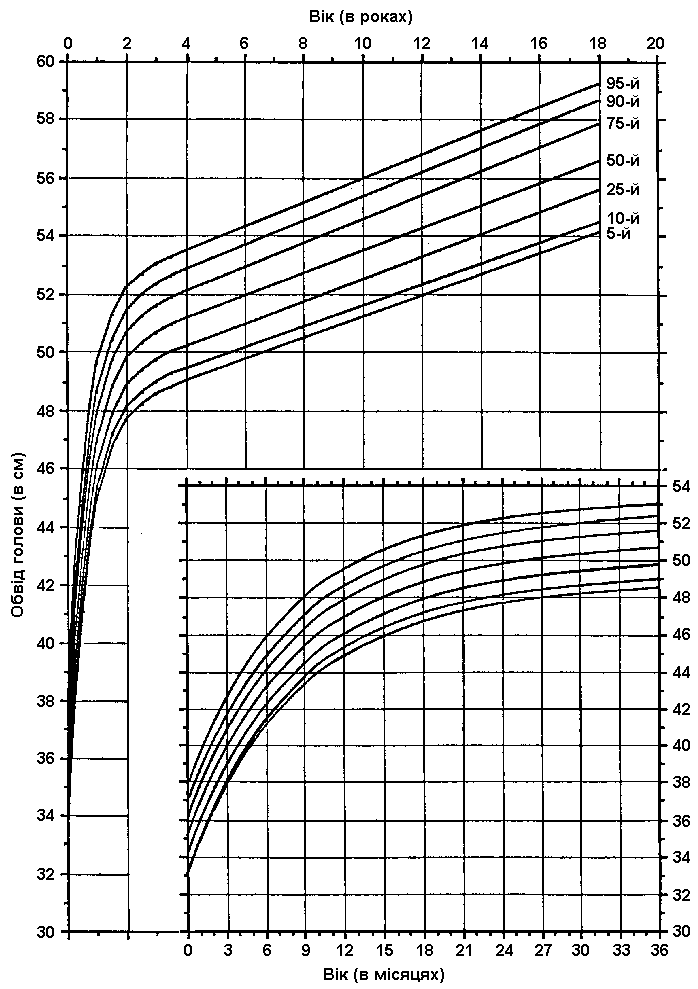
Вимірювання обводу голови проводилось у хлопчиків віком від народження до 18 років. Загалом в рамках довготривалої програми спостереження Фелс протягом 1930-1982 років було обстежено 888 пацієнтів, які проживали в південно-західній частині штату Огайо. Усі пацієнти належали до різних соціально-економічних груп. Roche AF, Mukherjee D, Guo S et al: Head circumference reference data: Birth to 18 years, Pediatrics 79:706, 1987.
Співвідношення росту та ваги у дівчат від 2 до 18 років
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”.
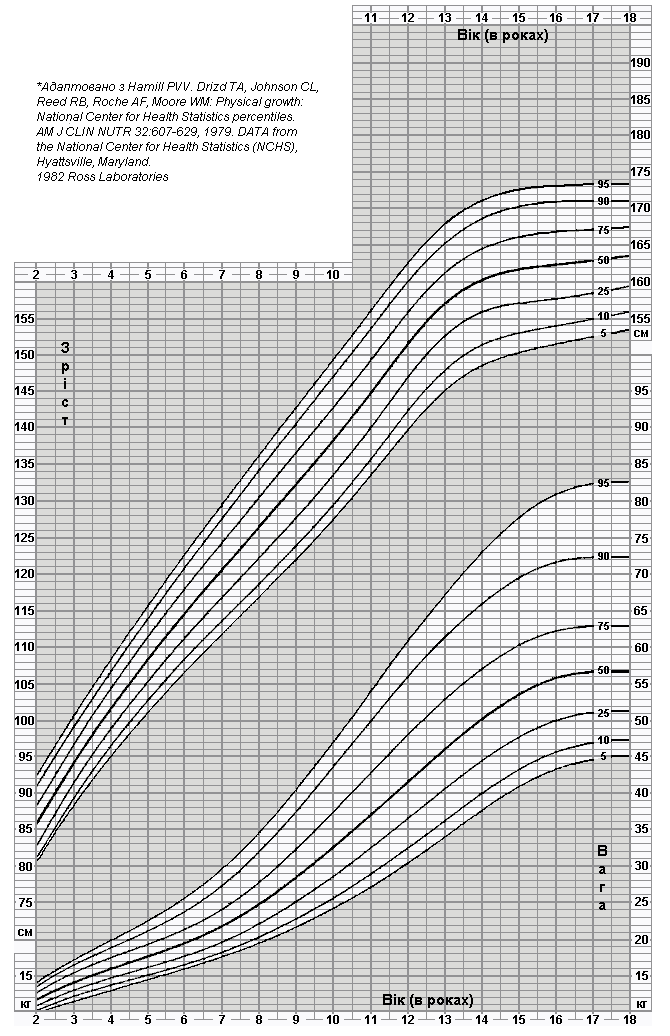
Дані таблиці підготовлено Національним центром медичної статистики на основі даних моніторингу фізичного розвитку і харчування за 1963-1975 роки. При цьому було обстежено понад 20000 пацієнтів. Вибірка охопила майже 70 мільйонів американських дітей у віці від 1 до 18 років. Hamill PVV, Drizd TA, Johnson CL, et al. Physical Growth: National Center for Health Statistics Percentile. Am J Clin Nutr 32: 607-629, 1979.
Співвідношення росту та ваги у дівчат від народження до 3 років
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”.

Дані таблиці підготовлено Національним центром медичної статистики на основі даних моніторингу фізичного розвитку і харчування за 1963-1975 роки. При цьому було обстежено понад 20000 пацієнтів. Вибірка охопила майже 70 мільйонів американських дітей у віці від 1 до 18 років. Hamill PVV, Drizd TA, Johnson CL, et al. Physical Growth: National Center for Health Statistics Percentile. Am J Clin Nutr 32: 607-629, 1979.
Співвідношення росту та ваги у хлопчиків від 2 до 18 років
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”.

Дані таблиці підготовлено Національним центром медичної статистики на основі даних моніторингу фізичного розвитку і харчування за 1963-1975 роки. При цьому було обстежено понад 20000 пацієнтів. Вибірка охопила майже 70 мільйонів американських дітей у віці від 1 до 18 років. Hamill PVV, Drizd TA, Johnson CL, et al. Physical Growth: National Center for Health Statistics Percentile. Am J Clin Nutr 32: 607-629, 1979.
Співвідношення росту та ваги у хлопчиків від народження до 3 років
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”.
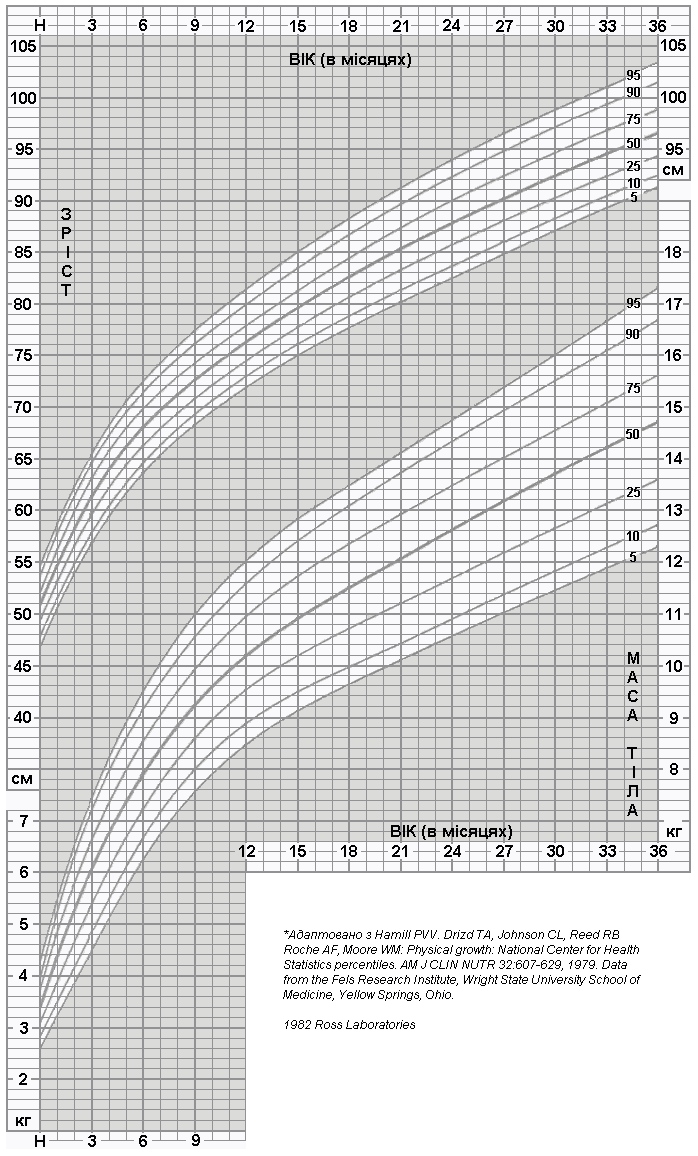
Дані таблиці підготовлено Національним центром медичної статистики на основі даних моніторингу фізичного розвитку і харчування за 1963-1975 роки. При цьому було обстежено понад 20000 пацієнтів. Вибірка охопила майже 70 мільйонів американських дітей у віці від 1 до 18 років. Hamill PVV, Drizd TA, Johnson CL, et al. Physical Growth: National Center for Health Statistics Percentile. Am J Clin Nutr 32: 607-629, 1979.
|
|
|
|
|
|






{kind=link}