
Serhiy
Хвороба Гоше
(Gaucher Disease)
Л.С. Євтушок
Зав. медико-генетичною консультацією
Рівненського клінічного лікувально-діагностичного центру
О.О. Михасюк
Спеціаліст з інформаційного забезпечення
Українсько-Американська Програма запобігання вродженим вадам розвитку
Вступ:
Хвороба Гоше отримала свою назву від імені французького лікаря Філіпа Чарльза Ернеста Гоше (Philip Gaucher), який в 1882 році вперше описав цю хворобу у пацієнта із збільшеною печінкою та селезінкою. В 1924 році німецький лікар виділив речовину, що містила жир, з селезінки пацієнтів з хворобою Гоше. Ще через 10 років інший французький лікар встановив, що цією речовиною є глюкоцереброзид – компонент, який знаходиться в оболонках червоних та білих кров’яних тілець.
Хвороба Гоше є аутосомно-рецесивною спадковою лізосомальною хворобою накопичення, що зумовлена мутацією гена b–глюкоцереброзидази. Дефіцит ферменту глюкоцереброзидази призводить до акумуляції глюкоцереброзиду. Глюкоцереброзидаза – це фермент, що розщеплює глюкоцереброзид, який знаходиться в кров’яних клітинах (макрофагах). В результаті дефіциту глюкоцереброзидази не відбувається нормального утворення клітин крові, а ті клітини, що утворюються, набувають специфічного вигляду – так званих “пінистих клітин” або клітин Гоше. Накопичення глюкоцереброзиду в органах та кістках може призвести до виникнення симптомів, які варіюють від легкої до важкої форми та проявляються в будь-якому віці від народження до старості. Існують три основних клінічних типи захворювання, які характеризуються наявністю або відсутністю неврологічної патології та прогресуванням хвороби:
- Тип I – доросла або неневрологічна форма.
- Тип ІІ – дитяча або гостра неврологічна форма.
- Тип ІІІ – підліткова.
Усі три типи успадковуються за аутосомно-рецесивним типом. Перший тип більш притаманний хворим єврейського походження (особливо євреям ашкеназі). Усі три типи хвороби мають одну спільну рису: прогресування.
Тип І хвороби Гоше зустрічається найчастіше, при ньому нервова система не уражена. Перебіг І типу може бути різним. У деяких випадках симптоми відсутні. Хворі ведуть нормальний спосіб життя, в той час, як інші відчувають значний дискомфорт та мають симптоми, що становлять загрозу для життя.
Типи ІІ і ІІІ є менш розповсюдженими і разом становлять менше 1% усіх хворих на хворобу Гоше. На відміну від І типу захворювання їм притаманно ураження нервової системи (при цьому при ІІІ типі воно є менш важким, ніж при ІІ). Середня тривалість життя хворих з ІІ та ІІІ типами хвороби Гоше становить 2-3 роки. Зазвичай смерть настає внаслідок важких порушень нервової системи.
Патофізіологія:
Глюкозилкерамід, накопичений глюколіпід, виділяється під час фагоцитозу та деградації старих лейкоцитів і, в меншій кількості, з еритроцитних мембран. Накопичення гліколіпідів спричиняє органомегалію та інколи легеневу інфільтрацію. Втрата нейрональних клітин в пацієнтів з ІІ та ІІІ типами хвороби ймовірно викликається накопиченням цитотоксичних гліколіпідів, глюкосфінгозину в мозку при важкому дефіциті активності кислої бета-глюкозидази.
Акумуляція глюкозилкерамідів в кістковому мозку, печінці, селезінці, легенях та інших органах призводить до панцитопенії, гепатоспленомегалії та дифузної інфільтрації легень. Прогресуюча інфільтрація клітинами Гоше кісткового мозку спричиняє потоншення, патологічні тріщини, біль у кістках, переломи кісток, остеопенію тощо.
Центральна нервова система (ЦНС) уражується лише у пацієнтів з ІІ або ІІІ типом захворювання. Важкість першого типу хвороби Гоше варіює від значного до легкого. В одних пацієнтів з раннього дитинства можуть спостерігатися усі прояви захворювання, тоді як інші випадки можуть залишатися асимптоматичними до 80 років.
При І типі хвороби Гоше характерними є виразні клінічні варіації. Ймовірно, обсяг залишкової ензиматичної активності визначає підтип та важкість прояву хвороби.
Молекулярна основа захворювання у 90-95% пацієнтів з хворобою Гоше, які мали єврейське походження, була визначена нещодавно. Також було зроблено опис мутацій генів серед хворих з інших етнічних груп.
Зв’язок між генотипом та фенотипом було помічено в багатьох випадках різних підтипів хвороби Гоше, це пояснює молекулярну основу різноманітності клінічних проявів.
Наприклад, при І типі хвороби Гоше пацієнти з гомозиготною мутацією N370S, мають тенденцію до більш пізнього прояву та відносно легкого перебігу ніж пацієнти з однією N370S алеллю або іншою мутованою алеллю.
Проте, різноманітність клінічних проявів хвороби Гоше не можна повністю пояснити лише мутаціями кислої бета-глюкозидази. Відомі випадки, коли в двох сибсів з ідентичними генотипами ступінь важкості значно відрізнявся.
Як і очікувалося, молекулярні порушення, які обумовлюють важкість проявів ІІ типу хвороби Гоше (інфантильного), при проведенні лабораторних досліджень виявляють незначну ензиматичну активність.
Як видно з нижчеподаної таблиці, на 100000 населення припадає лише 1 хворий. Серед євреїв ашкеназі (вихідців з країн Східної Європи) частота захворювання є вищою і сягає в середньому 1 на 850 осіб. Підвищена частота хвороби Гоше серед цієї групи населення призвела до неправильного уявлення про захворювання, як про “єврейську спадкову хворобу”. Насправді, на цю хворобу страждають особи будь-якої етнічної чи расової приналежності.
Частота:
(1 на 450–1500 серед євреїв ашкеназі) |
|||
Захворюваність/смертність:
- Тип 1 хвороби Гоше найчастіше проявляється у дитинстві (спленомегалія, панцитопенія, ураження скелету); клінічні прояви хвороби визначають важкість захворювання.
- Тип 2 (нейронопатичний) спричиняє швидко прогресуючу нейровегетативну хворобу та смерть в ранньому дитинстві.
- Тип 3 (підгостра нейронопатія) – це нейровегетативна хвороба, що прогресує не так швидко; смерть настає в дитинстві або ранньому підлітковому віці.
Успадкування:
Зазвичай у людини є 46 хромосом, включаючи дві, що визначають її стать (дві “Х” хромосоми у жінок, та одна “Х” і одна “Y” хромосома у чоловіків). Решта 22 пари хромосом називаються аутосомами. Ген ферменту глюкоцереброзидази знаходиться на одній аутосомній парі хромосом. Тому хвороба Гоше є аутосомно-рецесивним захворюванням. „Рецесивний” означає, що для розвитку хвороби людина повинна отримати дві мутованих копії гену, по одній від кожного з батьків. Тому усі три типи хвороби Гоше мають однакове поширення серед обох статей.
Людина з одним дефектним геном та одним нормальним геном глюкоцереброзидази є носієм хвороби Гоше. У носія хвороба не розвивається, оскільки один з двох генів глюкоцереброзидази є нормальним, тому виробляється достатня кількість ферменту, щоб не дати глюкоцереброзиду накопичуватися. Хоча носії хвороби Гоше не матимуть симптомів захворювання, існує вірогідність 50:50 того, що “ген Гоше” перейде до кожної дитини.
Якщо обидва батьки страждають на хворобу Гоше, то усі їх діти успадкують два “гени Гоше” і, відповідно, саму хворобу.
Дитина, в якої лише один з батьків є носієм, має 50% вірогідність самому стати носієм. Якщо ж обоє батьків є носіями хвороби Гоше, з кожною вагітністю існує 50% вірогідність того, що дитина успадкує один “ген Гоше” від кожного з батьків та буде носієм, або 25% ризику захворіти самій. Останнє також означає й те, що при кожній вагітності батьки-носії мають 75% вірогідність того, що їх дитина буде здоровою. Вірогідність успадкувати хворобу Гоше не залежить від того, чи була хворою дитина від попередньої вагітності.
Якщо тільки один з батьків має хворобу Гоше, а інший є здоровим і не є носієм, усі діти успадкують “ген Гоше” від хворого батька та стануть носіями. Проте, жоден з них не буде хворим.
Якщо один з батьків є лише носієм хвороби Гоше, тоді як інший є хворим, існує 50% вірогідність того, що дитина успадкує обидва мутованих гени і матиме хворобу. Також існує 50% вірогідність того, що дитина успадкує лише один хворий ген і стане носієм захворювання.
Родинна історія (родовід):
Оскільки хвороба Гоше успадковується за аутосомно-рецесивним типом, є цілком звичайним, що пробанд є першим хворим у родині. Тому необхідно дослідити родовід сім’ї для визначення того, чи є у хворого кровні сестри чи брати і чи вони не мають цього захворювання, а також для встановлення того, хто з них є носієм. Обстеження родини хворого також дасть можливість зробити мутаційний аналіз для визначення зразку генотип–фенотип (кореляція мутація-синдром).
Фенотипи хвороби Гоше:
Тип I
- З самого початку в усіх хворих на перший тип хвороби Гоше спостерігається хронічна втома, яка є вторинною до анемії; гепатомегалія з або без аномальних печінкових тестів; спленомегалія; біль у кістках або патологічні переломи; поширеними є великі синці, що є наслідком тромбоцитопенії.
- В декого з пацієнтів спостерігаються легеневі порушення або портальна гіпертензія.
- Більшість пацієнтів, в яких діагноз було встановлено протягом перших 5 років життя, мають більшу схильність до розвитку злоякісних захворювань.
- Пацієнти з менш вираженими проявами хвороби Гоше потрапляють в поле зору лікарів пізніше, в міру виникнення у них гематологічних порушень чи уражень скелету; інколи першим симптомом є спленомегалія.
- Вторинна до тромбоцитопенії кровотеча може проявлятися у вигляді носової кровотечі або синців.
- Клінічне ураження кісток, яке виникає в понад 20% пацієнтів з хворобою Гоше, може проявлятися у вигляді болю у кістках чи патологічних переломів.
- В більшості пацієнтів наявні радіологічні ознаки ураження скелету, включаючи колбоподібну деформацію Ерленмеєра периферичної ділянки стегна, що є наслідком ранніх змін у скелеті.
- В пацієнтів з симптоматичною хворобою кісток, в трубчастих кістках, ребрах та кістках тазу може розвинутися ураження, а також вже в ранньому віці може проявлятися остеосклероз.
- Кісткові кризи з сильним болем та розвитком пухлин виникають у пацієнтів із першим типом хвороби Гоше; вони часто помилково приймаються за синовіїт або остеомієліт, доки не з’являються інші симптоми.
- Спленомегалія є прогресуючою та швидко стає дуже значною. Діти з вираженою спленомегалією зазвичай мають коротку статуру.
Тип II
- Другий тип захворювання є досить рідкісним; він характеризується швидким нейродегенеративним перебігом з ураженням внутрішніх органів та загибеллю хворої дитини упродовж перших двох років життя.
- Хвороба проявляється в ранньому дитинстві; характерними рисами є підвищений м’язовий тонус, косоокість та органомегалія.
- Прогресуюча психомоторна деградація, що призводить до смерті, зазвичай спричиняється респіраторними ускладненнями.
Тип III
- Ця форма хвороби Гоше також проявляється в ранньому дитинстві або підлітковому віці.
- Окрім органомегалії та ураження кісток, наявними є серйозні неврологічні порушення.
- Розрізняють два підтипи третього типу хвороби Гоше – 3а та 3б. Різниця полягає в ступені важкості неврологічного ураження, наявності міотонії і розумової затримки (3а) та наявності ізольованого супрануклеарного паралічу зорового нерву (3б).
Клінічні прояви:
Фізичний огляд при першому типі хвороби Гоше зазвичай дозволяє виявити наявність гепатоспленомегалії. Спленомегалія може бути вираженою, навіть із зміщенням в сторону тазу. Коротка статура спостерігається в пацієнтів з органомегалією. Крім зазначених проявів у пацієнтів з ІІ та ІІІ типами хвороби спостерігаються затримка розвитку та аномальні результати неврологічних досліджень – підвищені сухожилкові рефлекси.
Спленомегалія. Накопичення клітин Гоше в селезінці викликає її збільшення (спленомегалію) та гіперактивність органу. Розміри селезінки можуть збільшуватися в 25 разів, внаслідок чого живіт виглядатиме як при ожирінні чи вагітності. Надмірна активність селезінки призводить до більш швидкого руйнування червоних кров’яних тілець (еритроцитів). Це призводить до анемії. Еритроцити несуть кисень з легенів в усі клітин організму. Оскільки у хворих з анемією не вистачає еритроцитів, їх тканини страждають від кисневої недостатності, що викликає швидку втому. Гіперактивність селезінки також може призвести до зниження кількості тромбоцитів в крові (тромбоцитопенію). Зменшення кількості тромбоцитів погіршує здатність зсідання крові. Це підвищує схильність до кровотеч та гематом. Тому у пацієнтів з хворобою Гоше набагато частіше за інших людей розвиваються важкі носові кровотечі та кровоточивість ясен. Менструації також можуть бути більш об’ємними та тривалими. Надмірна функціональна активність селезінки може призвести до зниження кількості білих кров’яних тілець (лейкоцитів).
Коливання кількості лейкоцитів в організмі є природнім, наприклад, при бактеріальній чи вірусній інфекції. Проте зниження кількості лейкоцитів, що виникає у відповідь на присутність клітин Гоше в селезінці, негативно впливає на здатність організму опиратися інфекціям. Нижче наведені основні клінічні прояви спленомегалії:
- В хворих спостерігається спленомегалія різного ступеню важкості – від 5 до 80 одиниць (фолдів) по відношенню до ваги тіла (приблизна нормальна вага селезінки становить 0,2% від загальної ваги тіла). Абсолютний розмір селезінки коливається від 300 грамів до 10 кг, сягаючи таким чином 25% від загальної ваги тіла.
- Збільшення селезінки в дітей з хворобою Гоше відбувається швидше, ніж в інших. Швидке збільшення селезінки у дорослих може наштовхнути лікаря на пошук поєднаних порушень, які здатні спричинити підвищення обміну гліколіпідів (наприклад, злоякісні гематологічні захворювання, імунна тромбоцитопенія, аутоімунна гемолітична анемія тощо).
- Горбкуватість поверхні селезінки може бути зумовленою ділянками екстрамедулярного гемопоезу, скупченням клітин Гоше або інфарктом. Більшість інфарктів селезінки є асимптоматичними, але субкапсулярні інфаркти можуть проявлятися у вигляді локалізованого болю в черевній порожнині.
Гепатомегалія. Клітини Гоше можуть накопичуватися в печінці. Результатом цього стає збільшення печінки у розмірах (цей стан називається гепатомегалією). Так, акумуляція клітин Гоше може викликати цироз, утворення рубців в печінці та розвиток інших типів печінкової дисфункції. В пацієнтів із гепатомегалією спостерігається підвищена схильність до утворення каменів в жовчному міхурі.
Гепатомегалія виникає більше, ніж в 50% пацієнтів з першим типом хвороби Гоше. При цьому печінка нормального розміру повинна становити 2,5% ваги тіла з середнім відхиленням в 1,75%.
Рівні глюкоцереброзидів в печінці коливаються від 23 до 389 одиниць вище нормального рівня.
Надмірне збільшення печінки зазвичай можна виявити за допомогою пальпації, особливо ділянки з нерівною поверхнею. Цироз і портальна гіпертензія не є характерними, проте спостерігаються в незначної кількості пацієнтів з хворобою Гоше.
Компресія синусоїдів клітинами Гоше може загострити портальну гіпертензію. Відомі летальні випадки внаслідок варикозної кровотечі.
Типовою рисою є незначне відхилення рівня ензимів печінки навіть в пацієнтів із порівняно легким ступенем хвороби Гоше. При цьому наявність жовтяниці або порушення функції гепатоцелюлярного синтезу є поганими ознаками. Жовтяниця в пацієнтів з хворобою Гоше зазвичай є результатом інфекції, розвитку хронічного гепатиту чи інколи навіть печінкової декомпенсації. Гемоліз визначається при наявності непоєднаної (неконьюгованої) гіпербілірубінемії.
Підвищення рівня сироваткового ферритину інколи спостерігається в пацієнтів з хворобою Гоше; при цьому рівень насиченості трансферину зазвичай є в межах норми. При проведенні біопсії печінки в синусоїді виявляються перевантажені гліколіпідами клітини Гоше, але гепатоцити не виявляють явного накопичення гліколіпідів, можливо внаслідок екскреції глюкоцереброзидів жовчним міхуром та того факту, що екзогенний обіг гліколіпідів регулюється мононуклеарними (одноядерними) фагоцитами.
Скелетні прояви. В більшості пацієнтів з хворобою Гоше відбувається залучення в патологічний процес кісток, яке зазвичай прогресує та погіршує загальний стан хворих.
Накопичення клітин Гоше в кістковому мозку може призвести до різноманітних уражень кісток. Це може бути обмеження притоку крові, що руйнує кісткову тканину (асептичний некроз) та призводить до постійного порушення рухливості суглобів. Крім того, може знижуватися маса кісток (остеопенія), порушуватися мінеральний обмін кальцію та фосфору, які є необхідними для збереження міцності та форми кісток. При цьому в кісткову тканину може проникати інфекція або кістки стають тоншими та більш слабкими, ніж нормальні. Це призводить до того, що переломи стають звичним ускладненням в пацієнтів з хворобою Гоше.
Залучення скелетної системи до патологічних процесів при даному захворюванні призводить до того, що в кістках утворюються аномальні ділянки ущільнення кісткової тканини (склероз) вздовж стрижня кістки або такі структурні зміни, як сплощення голівки стегнової кістки. Наприклад, в той час як для здорової кістки характерною є округла форма голівки, кістка, уражена хворобою Гоше, може мати розплющену форму біля закінчення голівки стегнової кістки. Таке явище носить назву деформації у вигляді колби Ерленмейєра, оскільки на рентгенологічному знімку кістка має схожу форму. Ця незвичність форми вказує на утворення нової кістки (процес ремоделювання кісток) у відповідь на присутність клітин Гоше в кістковому мозку.
Великі кризи відбуваються при виникненні гострої ішемії кісткової тканини з порушенням нормального кровотоку внаслідок стискання судин скупченнями клітин Гоше. Кісткові кризи характеризуються виникненням інтенсивного гострого болю (деякі пацієнти описують його як відчуття, схоже на “напад стенокардії в кістці”), який може тривати кілька годин або днів. Кісткові кризи викликаються розвитком набряку всередині та навколо кістки, що зменшує кровопостачання кістки (оклюзія судин).
Скелетні прояви при хворобі Гоше є багатогранними. Вони можуть коливатися від асимптоматичної колбоподібної деформації Ерленмейєра дистальної ділянки стегна до патологічних переломів, колапсу хребта та гострого больового кризу, який може помилково трактуватися як гострий остеомієліт.
Больові кісткові кризи настають внаслідок кісткового інфаркту, який призводить до остеосклерозу так само, як це відбувається при серпоподібно-клітинній анемії.
В дітей з хворобою Гоше гостре ураження стегна може неправильно трактуватись як хвороба Легга–Кальве–Пертеса, а ішемічний некроз стегна є найпоширенішим ускладненням.
Гематологічні ускладнення. Гематологічні прояви хвороби Гоше включають цитопенію та набуту коагулопатію, які спричиняються дефіцитом фактору ХІ. При цьому в осіб єврейського походження спостерігається генетичний дефіцит фактору ХІ, отож це може бути супутнім фактором при хворобі Гоше.
Коли в пацієнта із видаленою селезінкою виявляється цитопенія, це відображає підвищену інфільтрацію кісткового мозку клітинами Гоше. Порушення кісткового мозку та мієлофіброз виникають лише в незначної кількості таких пацієнтів.
Найпоширеніші імунологічні аномалії, притаманні пацієнтам з хворобою Гоше, включають: гіпергамаглобулінемію, дефіцит Т-лімфоцитів в селезінці та нерівномірний хемотаксис нейтрофілів.
Діагностика хвороби Гоше:
В цілому діагностика хвороби Гоше не є складною. Проте, оскільки можливі симптоми захворювання часто пов’язують з іншими, більш поширеними захворюваннями, хворому спочатку можуть встановити хибний діагноз. Наприклад, біль у суглобах можуть пов’язувати з артритом або “хворобою росту” замість хвороби Гоше; високий рівень лейкоцитів або низький рівень еритроцитів чи тромбоцитів можуть діагностувати як лейкемію.
Точний та визначений діагноз хвороби Гоше ставиться за допомогою аналізу крові, який дозволяє визначити активність ферменту. У здорових людей цей тест показує нормальну активність ферменту в лейкоцитах, у пацієнтів з хворобою Гоше активність ферменту значно знижена.
Загальне фізичне обстеження може включати загальний аналіз крові для підрахування еритроцитів, лейкоцитів та тромбоцитів. Але більш точним й специфічним тестом є ферментний аналіз. Він визначає та вимірює активність певних речовин, наприклад, ферментів в крові. Визначення глюкоцереброзидази в лейкоцитах або культурі фібробластів шкіри (клітин шкіри) встановлює діагноз хвороби Гоше.
Клінічні симптоми, що дозволяють припустити наявність в пацієнта хвороби Гоше, включають:
- анемію;
- збільшення селезінки та печінки;
- тромбоцитопенію;
- кісткову патологію (атрофію, склероз, некроз);
- в рідкісних випадках неврологічні порушення (горизонтальний парез зору, міоклонічну епілепсію).
Гематологічні показники:
- гемоглобін нижчий за норму (13-18 г/дл для чоловіків та 12-16 г/дл для жінок);
- тромбоцити нижче норми (150-350 нг/мл).
Вісцеральні показники:
- збільшена селезінка з вогнищами інфаркту;
- збільшена печінка з фіброзними ділянками.
Нижче поданий список можливих допоміжних лабораторних аналізів, які є корисними для встановлення точного діагнозу при хворобі Гоше та визначення її ступеня важкості:
- Активність кислої бета-глюкозидази: діагноз може бути підтверджений встановленням рівня активності кислої бета-глюкозидази в лейкоцитах периферичної крові. Якщо показник активності є меншим за 15 відсотків від середнього нормального показника, це є переконливим підтвердженням діагнозу. Зазвичай в гетерозигот спостерігається напівнормальна ензимна активність, але часом можуть реєструватися коливання в той чи інший бік на 20%.
- Генотип кислої бета-глюкозидази: молекулярна діагностика може бути корисною, особливо у випадках з пацієнтами ашкеназійського походження, в яких 4 типи мутацій в гені кислої бета-глюкозидази (N370S, 84GG, L444P, IVS 2+1) спричиняють майже 97% хвороби. Мутаційний аналіз має також значення для визначення прогресування хвороби.
- Загальний аналіз крові: дозволяє визначити ступінь цитопенії.
- Печінкові проби: незначне підвищення активності ензимів печінки є характерним для пацієнтів з хворобою Гоше, навіть у тих, що мають захворювання середньої важкості. Ознаками несприятливого прогнозу є наявність жовтяниці або порушення функції гепатоцелюлярного (гепатоклітинного) синтезу.
Радіологія:
- Ультрасонографія: УЗД органів черевної порожнини дозволяє встановити рівень органомегалії.
- Магнітно-ядерний резонанс (МЯР):
- МЯР є більш точним у визначенні розмірів внутрішніх органів.
- МЯР стегна є корисним для ранньої діагностики безсудинного некрозу.
- МЯР можна використовувати для встановлення ступеня інфільтрації кісткового мозку.
- Радіографія:
- Рентгенографія скелету може встановити скелетні прояви хвороби Гоше.
- Рентгенографія грудної клітки допоможе встановити наявність легеневих проявів захворювання.
Процедури:
- Біопсія кісткового мозку:
- Ця діагностична процедура може бути запропонованою для пошуку наявності класичних гліколіпід-наповнених макрофагів в пунктаті кісткового мозку. Проте псевдо-клітини Гоше були описані в пацієнтів з іншими порушеннями, такими як хронічна гранулоцитарна лейкемія, таласемія, множинна мієлома, хвороба Ходжкіна, плазмоцитоїдна лімфома, а також у хворих на СНІД.
- Аспірація кісткового мозку не повинна бути основним діагностичним тестом, оскільки аналіз ензимів крові є більш чутливим, специфічним та менш інвазивним.
- Біопсія печінки:
- Біопсія печінки проводиться лише в окремих випадках, коли наявною є гепатомегалія невизначеного генезу.
- В більшості пацієнтів біопсію печінки можна замінити іншими діагностичними тестами.
Гістологічні дослідження. При хворобі Гоше при аспірації кісткового мозку та біопсії печінки знаходять класичні гліколіпід-наповнені макрофаги. При біопсії печінки ці макрофаги спостерігаються в синусоїдах, проте в гепатоцитах немає ознак надмірного накопичення гліколіпідів в основному через екскрецію глюкоцереброзидів з жовчю, а також той факт, що екзогенний обмін гліколіпідів регулюється мононуклеарними фагоцитами. Недостатність гепатоцитів є постійною, але кількість випадків порушень функцій печінки у пацієнтів з хворобою Гоше є відносно малою.
Патологічною ознакою хвороби Гоше є наявність клітин Гоше в системі макрофаг-моноцитів, особливо в кістковому мозку. Ці клітини, які сягають 20-100 мікрометрів в діаметрі, мають характерний вигляд зім’ятого паперу, що є результатом внутрішньоцитоплазматичного відшарування від мембрани. Наявність клітин Гоше в кістковому мозку та / або інших тканинах дозволяє припустити наявність в пацієнта хвороби Гоше.
Медична допомога:
Ферментозамісна терапія при хворобі Гоше застосовується до більшості пацієнтів. Вони отримують рекомбінований ензим (іміглюцеразу [Церезим]). Кисла бета-глюкозидаза впливає на макрофаги і має високу ефективність в подоланні гематологічних проявів хвороби та внутрішніх органів, але майже не має позитивного впливу на скелетні прояви хвороби Гоше. Зазвичай лікування призначається наступним чином: раз на тиждень прийом великої дози препарату, тиждень перепочинку, знову лікування за тією ж схемою. Проте інколи пацієнтам призначають постійне безперервне лікування з вживанням середньої дози один раз на тиждень або малої дози три рази на тиждень. Позитивні результати лікування отримувались незалежно від режиму прийому медикаментів, отож так і не було встановлено якогось одного варіанту лікування.
Ферментозамісна терапія з використанням кислих бета-глюкозидів, що впливає на макрофаги, розрахована на лікування пацієнтів з І типом хвороби Гоше, в яких спостерігаються клінічні ознаки та симптоми, що викликають анемію, тромбоцитопенію, скелетні порушення або органомегалію. Оскільки важкість клінічних проявів надзвичайно мінлива, деяким пацієнтам дуже важко призначити відповідне лікування. В таких випадках варто застосувати досимптомне лікування для профілактики ішемічного некрозу.
В майбутньому пацієнтам з мутаціями генів, що зумовлюють несприятливий прогноз, також варто призначати переважно профілактичне лікування для попередження ускладнень ще до їх виникнення. Зазвичай, діти з хворобою Гоше, діагноз в яких встановлювався за клінічними ознаками, а не за даними родоводу, мають більш важкі прояви та потребують раннього медичного втручання.
Для більшості пацієнтів з хворобою Гоше лікування Церезимом або схожими препаратами має відбуватися під наглядом спеціаліста з метаболічних порушень та гематолога.
В 25% випадків після 6 місяців лікування відмічається зменшення розмірів печінки та селезінки. В пацієнтів з анемією гемоглобін піднімається до 1,5 г/дл протягом перших 4-6 місяців лікування. Протягом подальших 9-18 місяців гемоглобін піднімається ще на 1 г/дл. Рівень тромбоцитів змінюється більш повільно, подвоюючись десь наприкінці року від початку лікування. Але найповільніше відбуваються позитивні зміни в скелетних проявах хвороби Гоше. Хоча вже за рік після початку лікування зміни стають помітними, рентгенографічне підтвердження цьому можна отримати через значно довший проміжок часу.
Інші позитивні результати від ферментотерапії дітей з хворобою Гоше включають: стимуляцію росту і маси тіла, підвищення рухливості, покращання статевого розвитку та гіперметаболічного стану.
Ефективність ферментотерапії є мінливою та не пов’язана з генотипом, ступенем важкості хвороби, видаленням селезінки (спленектомією) чи віком. Низька ефективність лікування пов’язана з кількома факторами, такими як цироз, портальна гіпертензія, інфаркт та фіброз селезінки, легеневі ураження. Перелічені симптоми в поєднанні із злоякісними гематологічними проявами дають надзвичайно несприятливий прогноз для лікування.
Для того, щоб подолати перелічені труднощі можна порадити збільшити дозу та частоту прийому ензимів. Але у випадках, коли пацієнти мають декомпенсоване ураження печінки, ензимотерапія не завжди є ефективною. Крім того, в таких пацієнтів високим є ризик смерті від втрати крові внаслідок варикозної кровотечі. Слід також зазначити, що ферментотерапія ІІ та ІІІ типів хвороби Гоше є значно менш ефективною, але дослідження тривають.
Також потрібен постійний контроль за гематологічним станом хворих зі спленомегалією, оскільки вона може призвести до множинних порушень.
Пацієнти з гострим кістковим кризом потребують знеболювального.
Хірургічне втручання. В минулому для лікування пацієнтів з хворобою Гоше застосовувалась часткова або повна спленектомія. Оскільки зараз доступною є ферментозамісна терапія, застосування спленектомії в більшості пацієнтів можна уникнути.
Дорослі пацієнти з хворобою Гоше можуть потребувати протезування стегна через важке ураження скелету. Рекомендується перед протезуванням провести кількамісячний курс замісної ферментотерапії.
Додаткові консультації:
- Медико-генетична
- Гематолога
- Ортопеда
Дієта:
Під час досліджень не було доведено позитивного впливу дієти на стан здоров’я хворого.
Запобігання/профілактика:
Хвороба Гоше успадковується за аутосомно-рецесивним типом. Оскільки відомо, що особи єврейського походження мають більшу схильність до цієї хвороби, є очевидною необхідність проведення скринінгу серед цієї групи населення. В ідеалі програма скринінгу для виявлення носійства хвороби Гоше має поєднуватись з медико-генетичною службою для надання консультативної допомоги родинам, які відносяться до групи ризику.
Ускладнення:
- Кісткові кризи є вторинними по відношенню до інфарктів. Ішемічний некроз зустрічається надзвичайно часто.
- Розрив селезінки може відбуватися внаслідок травм.
- Цироз є досить рідкісним ускладненням.
- В рідкісних випадках легенева інфільтрація клітинами Гоше може проявлятися у вигляді симптомів ураження легень:
- Легенева інфільтрація або ущільнення легенів; це особливо притаманно пацієнтам з ІІ типом хвороби Гоше.
- Паренхіматозна інфільтрація з фіброзом була описана в дітей з ІІІ типом хвороби Гоше.
- Розширення внутрішньолегеневих судин при наявності чи відсутності портальної гіпертензії може спричиняти гіпоксію легень.
- Було описано кілька випадків легеневої гіпертензії в дорослих пацієнтів на фоні відсутності інфільтрації.
- Гематологічні симптоми, включаючи анемію, тромбоцитопенію та лейкемію, є найчастішими ускладненнями при хворобі Гоше.
- Другими за частотою йдуть імунологічні ускладнення, включаючи гіпергамаглобулінемію, недостатність Т-лімфоцитів в селезінці та порушення хемотаксису нейтрофілів.
Біль, пов’язаний з хворобою Гоше, може бути як помірним, так і важким. Справлятися з таким болем буває надзвичайно складно. Наприклад, в гіршому з випадків виникнення кісткових кризів він може обмежити нормальну діяльність, навіть легкі рухи стають хворобливими, порушується сон, і хворого доводиться госпіталізувати. Дорослі та діти з хворобою Гоше разом із своїм лікарем можуть намагатися віднайти кращі шляхи для подолання болю. В кожному окремому випадку вони можуть бути різними, оскільки не існує універсального знеболюючого засобу.
Боротьба з втомою:
Іншою проблемою, з якою стикаються деякі пацієнти з хворобою Гоше, є втома в результаті анемії та збільшення печінки і / або селезінки. Люди з важкою анемією можуть відчувати втому навіть після того, як вони проспали всю ніч. Деяким дітям бракує енергії та витривалості для того, щоб гратися з іншими дітьми. Їм важко залишатися уважними в класі або зосередитись на виконанні домашньої роботи. Для пацієнта з хворобою Гоше звичайні речі можуть вимагати надзвичайних зусиль. Однак більшість таких людей усвідомлює, що вони можуть вести нормальний спосіб життя, якщо оберуть правильний ритм та узгоджуватимуть свої плани з родиною, друзями, вчителями й оточуючими.
Вплив хвороби на зовнішність:
Зовнішній вигляд може бути великою проблемою для хворих із збільшеною селезінкою або печінкою та низьким ростом. Дитину чи дорослого можуть дражнити й знущатися з нього через те, що ця хвора людина є товстою, а інколи, навіть, схожою на вагітну.
Особливості відношення до дітей:
Батьки та вчителі мають схильність ставитись до хворої дитини, як до маленької. Це особливо стосується дітей з хворобою Гоше, оскільки внаслідок хвороби вони ростуть значно повільніше за своїх однолітків.
Збільшена печінка або селезінка, схильність до переломів кісток й інші можливі симптоми призводять до того, що діти з хворобою Гоше стають менш рухливими, вони не можуть займатися груповими видами спорту. Але вони можуть знайти для себе більш придатне заняття, наприклад плавання, їзда на велосипеді, танці.
У підлітків з цим захворюванням може з затримкою розпочатися період статевого дозрівання, хоча більшість з них наздоганяє своїх однолітків до 19-річного віку. Проте, підлітковий вік – це той час, коли уявлення про самого себе і те, як тебе сприймають однолітки, є надзвичайно важливим для стану психічного здоров’я. До того часу, коли хворі підлітки не наздоженуть своїх друзів, в них можуть виникати фізіологічні проблеми на протязі періоду емоційної неврівноваженості.
Дуже важливим є підбадьорювання цих дітей, особливо тих, чия хвороба має важку форму, розвиток в них інтересу до зовнішнього світу, діяльності та здоров’я. Діти часто компенсують ті речі, яких вони не можуть робити, перевагою в інших галузях. Лікарі та родини можуть діяти разом для визначення того, яка саме діяльність найбільш підходить тій чи іншій дитині з хворобою Гоше.
Переглянуто редакційною колегією I.B.I.S.: 06/03/2003
Голопрозенцефалія
(Holoprosencephaly)
Н. Ботаневич
Лікар-невролог
Рівненської дитячої обласної клінічної лікарні
Включення:
Аріненцефалія, циклопія, голотеленцефалія.
Виключення:
Апрозенцефалія, ателенцефалія, гідраненцефалія, аріненцефалія без голопрозенцефалії.
Визначення:
Голопрозенцефалія – група вад розвитку мозку, що викликані порушенням формування півкуль головного мозку та поділу діенцефалона та теленцефалона.
Асоційовані вади:
Мікроцефалія, лицеві фенотипи (циклопія, етмоцефалія, цебоцефалія,) плюс трігоноцефалія, аріненцефалія (агенезія нюхових цибулин), гіпотелоризм, латеральна розщілина губи, агнатія.
Класифікація:
- Рання, безчасткова (алобарна);
- Хронологічно пізніша, напівчасткова (семілобарна);
- Часткова (лобарна).
Алобарна голопрозенцефалія. Характерна відсутність поділу мозку на 2 півкулі, практично повна відсутність задніх відділів мозку та диспластичність передніх відділів, несформованість нормальної шлуночкової системи (є тільки один шлуночок), несформованість або аномальне формування підкоркових утворень. Часто мають місце аномалії зорових нервів та очних яблук, нюхових цибулин і нюхових трактів.
Семілобарна голопрозенцефалія. Частковий поділ коркового плаща на дві півкулі, мозолясте тіло звичайно відсутнє, як і рінецефалон (нюховий відділ мозку), основні вузли й зорові горби сполучаються в одну тканинну масу.
Лобарна голопрозенцефалія. Наявність диспластичних серединних структур мозку (мозолясте тіло, серпоподібний відросток твердої мозкової оболонки), відсутність прозорої перетинки, гіпоплазія лобних частин.
Критерії діагнозу: нейрорадіологічні ознаки (комп’ютерна томографія, МРТ).
Клінічна характеристика:
Алобарна голопрозенцефалія. Найбільш характерною ознакою є черепно-лицеві дизморфії – у важких випадках це єдиний шлуночок мозку, цебоцефалія та циклопія, в помірних – гіпоплазія переважно середньої третини обличчя, гіпотелоризм, серединна розщілина губи та піднебіння, лицева розщілина, гіпоплазія носа (агенезія кісток носа), відсутність фільтру. Виражена черепно-лицева дизморфія зазвичай поєднується з різноманітними пароксизмальними порушеннями: судомами, нападами апное, пойкілотермією. Практично в усіх пацієнтів відмічається виражена затримка нервово-психічного розвитку. Приблизно в 75% випадків алобарна голопрозенцефалія поєднується з множинними аномаліями внутрішніх органів та інших систем (органу зору, нейроендокринної (відсутність гіпофізу, гіпоплазія наднирників, гіпоглікемія) та кістково-суглобової систем, мікропеніс).
Семілобарна голопрозенцефалія. Характеризується черепно-лицевою дизморфією у вигляді гіпотелоризму, плаского носа, відсутністю перегородки носа, розщілиною губи та піднебіння. В окремих випадках лише мінімальні аномалії обличчя. Можливі також судоми, затримка нервово-психічного розвитку.
Лобарна голопрозенцефалія. Характеризується порушенням зору, нейроендокринною дизфункцією та затримкою нервово-психічного розвитку.
Дані лабораторно-інструментальних досліджень:
- Нейросонографія – найбільш чітко діагностується алобарна голопрозенцефалія. Відмічаються відсутність міжпівкульної щілини та серпоподібного відростка твердої мозкової оболонки, єдиний шлуночок великого розміру.
- Ехографія.
- Алобарна – відсутність серединного М-ехо; відсутність зображення порожнини прозорої перетинки: наявність однієї серединно розташованої шлуночкової порожнини; зорові бугри злиті в єдиний утвір; мозолясте тіло відсутнє; структури стовбура та мозочок збережені.
- Семілобарна – відсутність зображення порожнини прозорої перетинки; серединне М-ехо простежується тільки в задніх відділах; бокові шлуночки злиті в ділянці передніх рогів та тіл, задні роги розділені та розходяться у відповідні півкулі; зорові горби злиті в єдиний утвір; ІІІ шлуночок рудиментарний або відсутній; мозолисте тіло відсутнє; структури стовбура та мозочок збережені.
- Лобарна – відсутність зображення порожнини прозорої перетинки; серединне М-ехо простежується на всьому протязі; бокові шлуночки з’єднані в ділянці передніх рогів, тіла та задні роги розмежовані та розходяться у відповідні півкулі; зорові горби мають симетричне розташування; ІІІ шлуночок диференційований, структури мозку та мозочок без особливостей.
- Нейрорадіологічна діагностика: КТ, МРТ.
Частота виникнення:
1:13000 новонароджених. У спонтанно абортованих ембріонів вона в 50 разів вища, ніж в загальній популяції новонароджених.
Співвідношення статей: Ч1 : Ж1.
Вік прояву: пренатальний період (за даними УЗД) та відразу після народження.
Етіологія:
Описані сімейні випадки як з аутосомно-рецесивним (сімейна алобарна голопрозенцефалія, ГПЕ1), так і з аутосомно-домінантним типами успадкування: ГПЕ2 – аутосомно-домінантна (del 2p21), ГПЕ3 – аутосомно-домінантна (7q36), ГПЕ4 – аутосомно-домінантна (14q11.1-q13). Тому батьків уражених дітей слід перевірити на наявність легких симптомів, таких як єдиний центральний різець і відсутність носового хряща. У більшості родин з аутосомно-домінантною голопрозенцефалією цей ген розташований на 7q36 і позначений НРЕЗ. Зрештою, голопрозенцефалія спостерігалась у дітей матерів, що хворіли на цукровий діабет, та при синдромі Меккеля-Грубера. Голопрозенцефалія нерідко відмічається при цілій низці генетично-детермінованих та хромосомних синдромів:
- Голопрозенцефалія семілобарна з краніосиностозом (аутомно-рецесивна);
- Голопрозенцефалія в поєднанні із акінезією/ гіпокінезією плода (Х-зчеплена);
- del(2)(p21);
- dup(3 pter);
- del(7)(q36);
- del(13q);
- del(18p);
- del(21)(q22.3);
- Синдром Меккеля;
- Трисомія 13;
- Кільцева хромосома 13;
- Трисомія 15;
- Трисомія 18;
- Делеція хромосоми 18;
- Кільцева хромосома 18.
Патогенез:
Протягом третього тижня фетального розвитку прехордальна мезодерма мігрує в ділянку, що знаходиться перед хордою (спинною струною) і є дуже важливою у розвитку середньої третини обличчя. В той же час вона відіграє індуктивну роль в морфогенезі переднього мозку. Наслідки дефекту прехордальної мезодерми варіюють від ступеня недорозвиненості середньої третини обличчя, особливо серединного носового відростка до незавершеного морфогенезу прозенцефалону. Циклопія являє собою важке порушення раннього розвитку середньої третини обличчя; очі зростаються, нюхова плакода зливається в один трубкоподібний хоботок (пробошизис) вище ока, решітчаста та інші кісткові структури середньої лінії обличчя відсутні. Разом з циклопією не відбувається поділ клітин прозенцефалона, зростає незавершеність морфогенезу. Менш важкі аномалії проявляються у гіпотелоризмі та різних ступенях аномалій середньої третини обличчя, а також у недостатньому розвиткові прозенцефалона, що є більш характерно, ніж циклопія та часто включає розщілини губи та піднебіння. Важливою клінічною ознакою є недорозвиненість структур середньої лінії обличчя, таких, як гіпотелоризм чи відсутність фільтру або носової перегородки, що передбачає можливість серйозного порушення розвитку головного мозку та його функціонування.
Лікування:
Симптоматичне. У тих випадках, коли є судоми, призначається протисудомна терапія.
Прогноз:
Більшість дітей з алобарною голопрозенцефалією нежиттєздатні. Вони або народжуються мертвими, або гинуть в перші місяці життя. При семілобарній формі прогноз для життя більш сприятливий, однак частота порушень нервово-психічного розвитку достатньо висока.
Запобігання: генетичне консультування, пренатальна діагностика.
Номер з каталогу МІМ:
- 601370 Holoprosencephaly, semilobar, with Craniosynostosis
- 236100 Holoprosencephaly
- 157170 Holoprosencephaly 2; HPE2
- 142945 Holoprosencephaly 3; HPE3
- 142946 Holoprosencephaly 4; HPE4
- 306990 Holoprosencephaly with fetal akinesia/hypokinesia sequence
Література:
- Козлова С.И., Демикова Н.С., Семанова Е., Блинникова О.Е. Наследственные синдромы и медико–генетическое консультирование.- М.: Практика, 1996.- C. 88-89.
- Садлер Т.В. Медична ембріологія за Лангманом.- Львів: Наутілус, 2001.- C. 460-461.
- Темина П.А., Казанцев А.З. Наследственные нарушения нервно-психического развития детей.- М.: Медицина, 2001.- C. 372-374.
- Jones K.L. Smith’s Recognizable Patterns of Human Malformation. W.B.Saunders Company, Philadelphia. 1997:605-608.
- Warkany J. Congenital Malformations. Notes and Comments. Year Book Publisher, Chicago. 1981:201-202.
Переглянуто редакційною колегією I.B.I.S.: 24/12/2002
Обвід голови при народженні та гестаційний вік (1610 метрів над рівнем моря та вище)
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”.
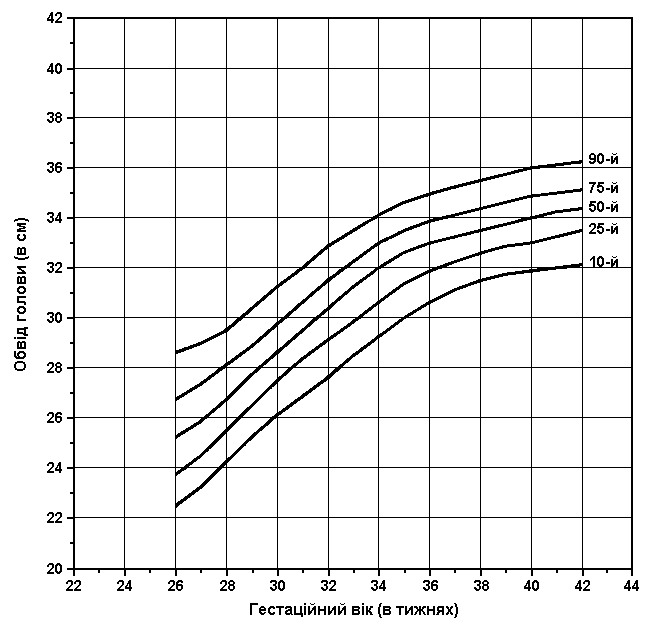
Значення 10, 25, 75 та 90 перцентилів обводу голови згідно з Lubchenco та співавт. (Pediatrics 37: 403, 1966). Дані отримані при огляді 4720 малюків, народжених в Денвері, Колорадо (1610 метрів над рівнем моря) у період між 1948 та 1961 роками. До уваги брались лише живонароджені діти без важких патологічних станів. Новонароджені латино-американського походження становили 30%.
Зріст при народженні та гестаційний вік (1610 метрів над рівнем моря та вище)
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”.

На графік нанесено 10-й, 25-й, 50-й, 70-й та 90-й перцентилі довжини тіла при народженні відповідно до гестаційного (менструального) віку. Дані отримані при обстеженні 7827 живонароджених дітей 24-42 тижнів гестації. Також включені дані про білих дітей та дітей латино-американського походження з Денвера, Колорадо (приблизно 1610 м вище рівня моря), народжених у період між 1948 та 1961 роками. До уваги не брались дані про дітей, молодших 26 тижнів та старших 42 тижнів гестації. Виключені також діти не європеоідної раси, великі для гестаційного віку та діти з очевидними аномаліями. Усі вимірювання виконувались протягом перших 24 годин життя. Lubchenco LO et al: Pediatrics 37: 403, 1966.
Вага при народженні та гестаційний вік (1610 метрів над рівнем моря та вище)
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”.
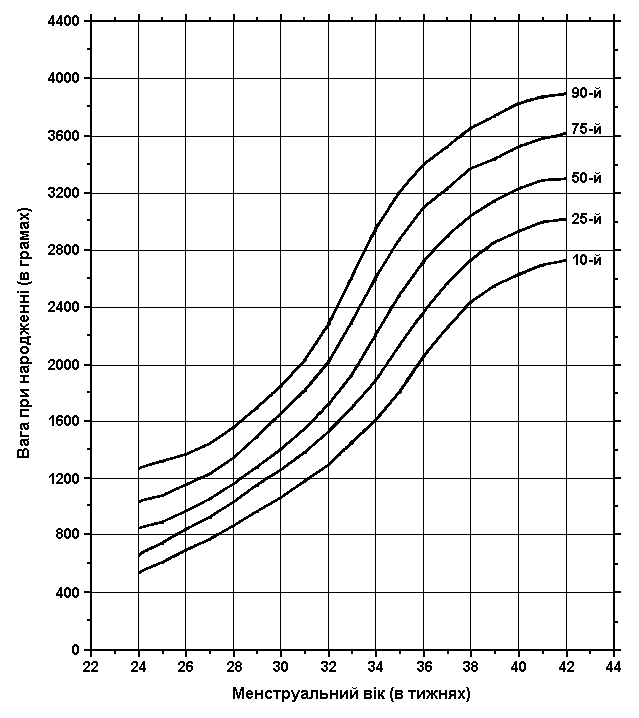
На графік нанесено 10-й, 25-й, 50-й, 70-й та 90-й перцентилі ваги при народженні відповідно до гестаційного (менструального) віку. Дані отримані при обстеженні 7827 живонароджених дітей 24-42 тижнів гестації. Також включені дані про білих дітей та дітей латино-американського походження з Денвера, Колорадо (приблизно 1610 м вище рівня моря), народжених у період між 1948 та 1961 роками. До уваги не брались дані про дітей, молодших 26 тижнів та старших 42 тижнів гестації. Виключені також діти не європеоідної раси, великі для гестаційного віку та діти з очевидними аномаліями. Усі вимірювання виконувались протягом перших 24 годин життя. Lubchenco LO et al: Pediatrics 37: 403, 1966.
Прикріплення та довжина пуповини
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”.
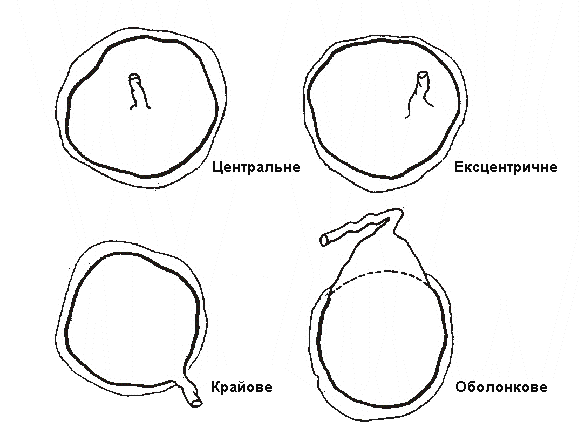
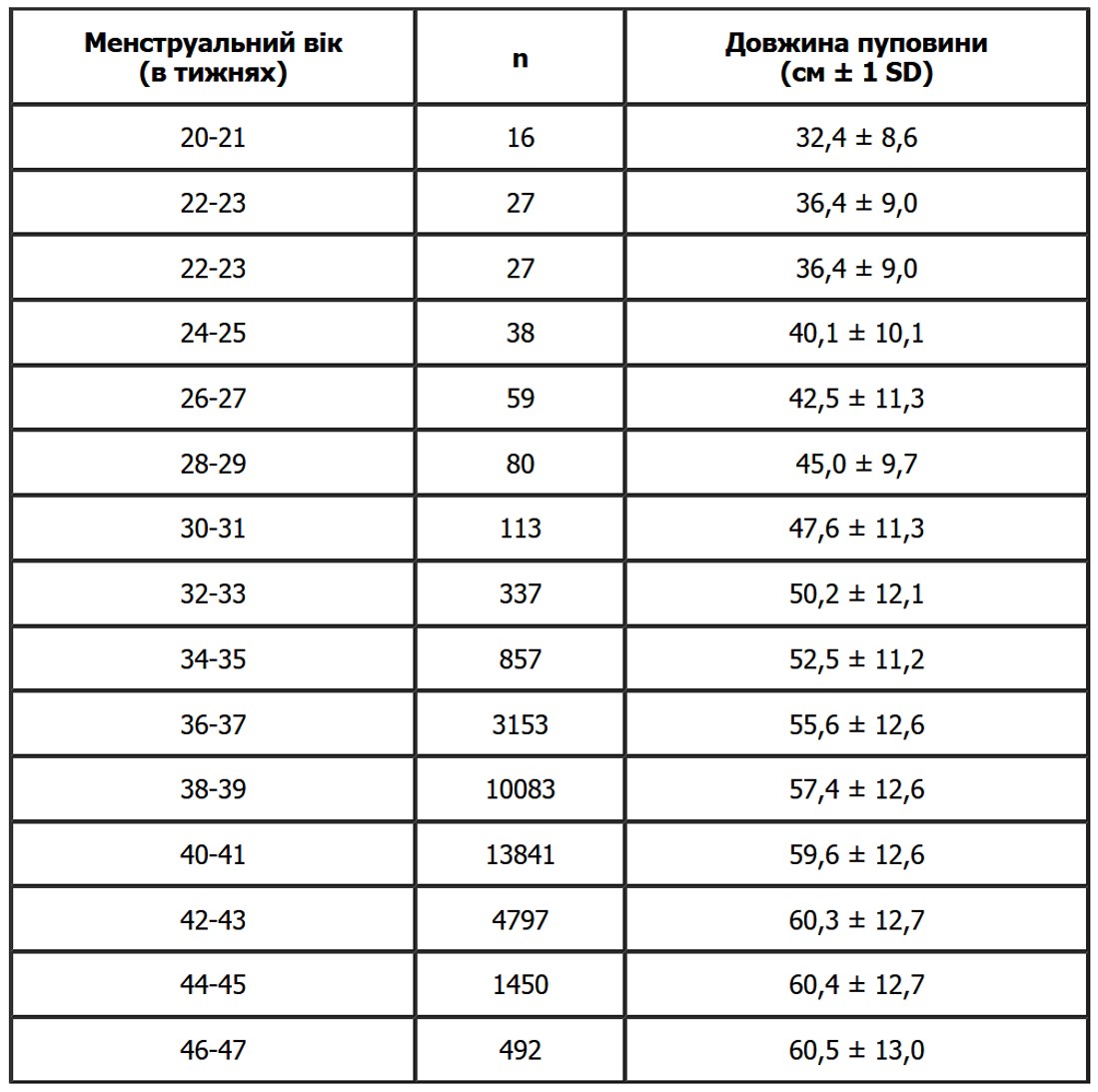
Довжина пуповини у 35779 новонароджених з одноплідних вагітностей за “Спільним перинатальним дослідженням”. Були виключені випадки з важкими вродженими вадами, випадки відшарування плаценти, розриву і травми пуповини. За Naеуе (J Pediatr 107: 278, 1985).
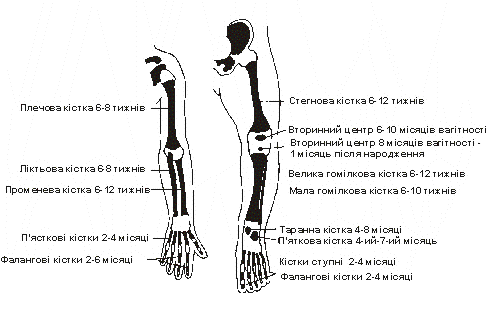
Осифікація
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”.
Виникнення центрів первинної осифікації згідно Potter and Craig (Pathology of the Fetus and the Infant, ed. 3, Year Book Medical Publishers, 1975). Фактично, усі центри первинної осифікації виникають ще до того, як плід стає життєздатним. Виключення становлять: п’ясткові та передплюсневі кістки, під’язикова кістка, лобкова кістка, куприк та середні фаланги пальців ніг. Малюнок взятий з книги Caffey (Pediatric X-Ray Diagnosis, ed. 6, Year Book Medical Publishers, 1972).

Скринінг глухоти
(Hearing Screening)
Н.С. Шкапій
Спеціаліст з інформаційного забезпечення
Українсько-Американська Програма запобігання вродженим вадам розвитку
“Щодня з пологових будинків Америки виписуються 22 малюки, які не мають можливості почути навіть адресовані їм ніжні слова батьків”,- говорить Елізабет Фостер, директор американської Національної Компанії “За здоровий слух”.
Що таке глухота?
Цим терміном в медицині позначають будь-яке виражене порушення слуху.
Як часто зустрічаються випадки порушення слуху серед населення?
Значна втрата слуху є досить розповсюдженою хворобою, в тому числі вродженою. Якщо захворювання не діагностоване вчасно, то це може призвести до порушення пізнавального розвитку та мови. Приблизно 3 із 1000 немовлят народжуються із значною втратою слуху, а у сім’ях, де випадки глухоти вже траплялись, вроджена глухота зустрічається майже у 4% новонароджених.
Якою може бути глухота?
З медичної точки зору глухота та значна втрата слуху можуть бути різних видів. Наприклад, розрізняють глухоту вроджену та набуту. Перша категорія порушення слуху, зазвичай, присутня при народженні, друга ж розвивається згодом.
За часом появи глухота характеризується термінами “прелінгвальна” та “постлінгвальна”. Прелінгвальні порушення слуху та глухоту встановлюють у перші 3 роки життя, тобто до початку появи мовлення. Зрозуміло, що вроджені вади слуху завжди будуть “прелінгвальними”. Якщо ж послаблення слуху виявилось після того, як дитина оволоділа мовленням, такі порушення називають “постлінгвальними”.
В залежності від того, який з фізіологічних аспектів слухового процесу пошкоджений у дитини, розрізняють три типи втрати слуху та глухоти. Якщо страждає процес звукопроведення – то порушення слуху називають кондуктивним. Якщо ж порушене сприйняття звуку, то глухоту називають сенсоневральною. Порушене проведення та сприйняття звуку називається змішаним.
Розрізняють глухоту спадкову та неспадкову. Різниця між ними полягає в тому, що перша передається від старшого покоління родини до молодшого, а друга – ні. Спадкова глухота має ще і свої власні форми – синдромні, несиндромні, із різним типом успадкування: аутосомно – рецесивним, домінантним та зчепленим із статтю, мітохондріальним та інші.
Порушення слуху можуть бути також повними та частковими – залежно від частот звуку, сприйняття яких ускладнене. В залежності від “поведінки” хвороби з часом – прогресуючі та непрогресуючі. Бувають гострі та хронічні форми, однобічні та двобічні, симетричні та асиметричні тощо.
З усього вище викладеного зрозуміло, що глухота є серйозною медичною та соціальною проблемою.
Як виявляється глухота?
Зазвичай, глухоту виявляють батьки, звертаючи увагу на неадекватну реакцію дитини на звернення до неї. Наступний крок – консультація та обстеження спеціаліста – сурдолога. Як правило, встановлення діагнозу є запізнілим. Одним з найбільш актуальних питань сучасної аудіології є вдосконалення методів діагностики порушень слуху. Успіхи в цьому напрямку, перш за все, визначаються своєчасністю встановлення діагнозу і, як наслідок, ефективністю як лікування, так і реабілітації хворих із порушеннями слуху. Рання діагностика втрати слуху може запобігти виникненню проблем з розвитком дитини. Саме тому в ряді країн із високим рівнем медичного обслуговування була запроваджена програма скринінгу глухоти усіх без виключення новонароджених.
Що таке скринінг глухоти?
Для встановлення порушень слуху застосовують так звану програму скринінгу (від англ. “screening” – просіювання), яка передбачає тестування усіх дітей на предмет втрати чи порушень слуху. Скринінг, що базується лише на реєстрації сімей, які належать до групи підвищеного ризику (наявність хвороби в родині), може визначити лише приблизно 50% новонароджених із значною вродженою втратою слуху. Виявлення втрати слуху батьками дитини чи лікарем часто є запізнілим. Скринінгове обстеження, проведене вчасно, “відсіває” дітей із вадами слуху. Це дозволяє на ранніх термінах розвитку хвороби направити малюків на більш детальне обстеження до спеціаліста – сурдолога.
Чому необхідно проводити скринінг глухоти?
Згідно даним, якими володіють американські спеціалісти, вроджені порушення слуху далеко не завжди діагностуються вчасно – найчастіше проходить кілька місяців, а іноді і років, перед тим, як такі діти потрапляють на прийом до сурдолога, де і виявляються порушення слуху та глухота. Зазвичай це призводить до неприпустимо пізнього початку слухо – мовної реабілітації і, відповідно, відставанню дитини в нервово – психічному розвитку.
Успішне виконання програми скринінгу дозволяє починати процес реабілітації глухих та малюків з проблемами слуху практично з народження. В цьому випадку соціальна адаптація таких дітей в суспільстві протікає значно успішніше, ніж при пізній діагностиці та пізньому початку лікувальних заходів.
На думку спеціалістів, не менш важливу роль в малому обсязі скринінгу малюків відіграє і недостатня обізнаність батьків: переважна більшість просто не знають про існування недорого та надійного методу перевірки слуху маленьких дітей. Тому слід посилити пропагандистську діяльність, з метою пояснення необхідності ранньої оцінки слуху малюків та домагатись офіційного визнання неонатального скринінгу глухоти медичною послугою, яка б в обов’язковому порядку надавалась виключно усім новонародженим.
Які вимоги до успішної реалізації скринінгової програми?
Для ефективної реалізації програми скринінгу, тест для виявлення пошкоджень слуху повинен бути простим у використані та мати високий рівень чутливості. Вчасне скринінгове обстеження із встановленням діагнозу може запобігти виникненню багатьох проблем із здоров’ям малюка та, що найголовніше, уникнути такого не менш серйозного захворювання, як німота. Тому батьки та лікарі, працюючи разом, відіграють важливу роль у запобіганні таких ускладнень.
Які методи застосовують для скринінгу глухоти?
Особливе місце у діагностиці стану слухового аналізатора займають об’єктивні методи дослідження. І найбільш перспективними з них на сьогоднішній день вважаються методи реєстрації і аналізу отоакустичної емісії та метод слухової реакції стовбура мозку (СРСМ). Обидва методи можна застосовувати як окремо, так і в комплексі один з одним. Отоакустична емісія являє собою надзвичайно слабкі звукові коливання, що генеруються внутрішнім вухом через кілька мілісекунд після сприйняття короткого звукового сигналу, які можуть бути зареєстровані в зовнішньому слуховому проході за допомогою надчутливого мікрофону. За методом слухової реакції стовбура мозку вимірюються електроенцефалографічні хвилі, що виникають у відповідь на звукове подразнення через 3 електроди, розташовані на голові дитини. Найкращий результат можна отримати, якщо застосовувати ці два методи разом. Друге скринінгове обстеження, проведене через певний час, може підтвердити чи спростувати поставлений діагноз. Його можна провести безпосередньо перед випискою або протягом першого місяця життя дитини.
Переглянуто редакційною колегією I.B.I.S.: 11/06/2002
Хвороба Гіршпрунга
(Hirschsprung Disease)
О.О. Михасюк
Спеціаліст з інформаційного забезпечення
Українсько-Американська Програма запобігання вродженим вадам розвитку
Загальні відомості:
Хвороба Гіршпрунга отримала свою назву на честь датського вченого, який вперше описав це захворювання. Це вроджена вада, при якій порушується іннервація товстої кишки. В результаті цього частина товстої кишки перестає нормально функціонувати: утруднюються або повністю припиняються перистальтичні рухи кишківника, дитина страждає постійними закрепами. Першим симптомом захворювання у новонароджених є затримка меконію (первинного стільця), проте проноси також можуть бути одним з симптомів хвороби Гіршпрунга. Подразнення прямої кишки іноді тимчасово стимулює перистальтику, виділення меконію та газів, проте не усуває основної причини захворювання.
Товста кишка над ділянкою, яка не має іннервації, значно збільшується в об’ємі (у 2-3 рази) в порівнянні з нормальною. Такий стан називається мегаколон (велика товста кишка). Сам мегаколон може в свою чергу ускладнюватися ентероколітом (запаленням кишківника). Поступово тиск в товстій кишці зростає, в її стінках порушується всмоктування і вона починає виділяти більше рідини, ніж поглинає. Це може спричинити пронос та подальше зневоднення. Подібні стани самі по собі не зникають і вимагають обов’язкового лікування.
Іноді симптоми хвороби Гіршпрунга проявляються лише у віці 6-12 місяців, коли виникають постійні закрепи, надлишкове газоутворення та значне збільшення кишківника. При цьому, зазвичай, спостерігаються незначні та водянисті випорожнення. Самопочуття дитини може бути нормальним, проте частіше дитина відстає в розвитку та часто хворіє. При важкому перебігу хвороби характерні постійні закрепи, напади різкого болю в животі, температурна реакція, розвивається анемія.
Частота поширення:
За поширеністю, хвороба Гіршпрунга займає друге місце після пілоростенозу. Ця вроджена вада зустрічається в 1 з 10000 новонароджених і призводить до порушення прохідності шлунково-кишкового тракту. Є думка, що частота хвороби є значно вищою, але внаслідок того, що діти гинуть ще в неонатальному періоді від кишкових інфекцій, які виникають через ускладнення анатомічних дефектів товстої кишки, хвороба Гіршпрунга не діагностується. В 10% випадків хвороба Гіршпрунга зустрічається у рідних братів та сестер або в наступних поколіннях однієї сім’ї. У хлопчиків хвороба спостерігається в 4 рази частіше, ніж в дівчат. Без своєчасного хірургічного втручання хвороба Гіршпрунга закінчується смертю.
Причини:
Новонароджені не можуть самостійно звільнитися від меконію (первинного стільця) доки не проведеться подразнення анусу під час пальцевого обстеження. Згодом при хворобі Гіршпрунга дитина страждає закрепами або проносами. В кишківнику утворюється велика кількість газів, що часто супроводжується голосним гурчанням та випинанням кишківника. У старших дітей, в яких товста кишка вже збільшилась у розмірах, з’являються болі в животі. При цьому дитина має поганий апетит, втрачає вагу, відстає у фізичному розвитку, стає дратівливою.
Діагноз:
Під час огляду хворої дитини зазвичай виявляють випинання кишківника. При обстеженні прямої кишки відмічається відсутність в ній звичайного вмісту (при закрепах, спричинених іншими хворобами, пряма кишка заповнюється каловими масами). Лікар має призначити рентгенівське обстеження товстої кишки, після чого робиться вимір тиску в прямій кишці. Для підтвердження діагнозу проводиться біопсія стінки товстої кишки. Відсутність нервових клітин підтверджує припущення щодо хвороби Гіршпрунга.
Ускладнення:
Закрепи можуть спостерігатися при органічних ураженнях товстої кишки. При хворобі Гіршпрунга вони пов’язані з наявністю звуженої (агангліонарної) зони в дистальному відділі. Гістологічно це проявляється відсутністю клітин в ауербахівському нервовому сплетінні кишківника.
Клініка ентероколіту при хворобі Гіршпрунга залишається маловивченою, а тому й маловідомою для широкої аудиторії педіатрів та дитячих хірургів. При цьому в клініках дитячої хірургії третього рівня досі спостерігаються випадки хвороби Гіршпрунга з потенційно летальним її ускладненням – ентероколітом – у дітей 12 і навіть 14 років. Таким чином, патологія залишається недіагностованою багато років до та часто після оперативного лікування.
Раптовий розвиток важкого запального процесу в кишківнику (ентероколіт), який виникає без попередніх симптомів і може закінчитись смертю пацієнта, є найпоширенішим ускладненням хвороби Гіршпрунга. Якщо своєчасно не проведене зниження внутрішньокишкового тиску хірургічним шляхом та антибактеріальна коригуюча терапія, то смерть може настати через поширення інфекції та розвиток сепсису. Серед більш старших дітей найпоширенішим ускладненням є порушення фізичного розвитку.
Лікування:
Усі діти з хворобою Гіршпрунга повинні проходити стаціонарне лікування. Частина кишки, яка немає іннервації, видаляється хірургічним шляхом, а здорові краї з’єднуються.
Якщо у новонародженого з хворобою Гіршпрунга виник гострий запальний процес в кишківнику (ентероколіт), то найчастіше надійним засобом першої допомоги для оживлення роздутої кишки є тимчасова колостомія, при якій частина кишки виводиться назовні. Калові маси збираються в закріплений до зовнішньої черевної стінки приймач. Через кілька місяців при повторній операції видаляється частина кишки або вся уражена товста кишка, а тонка кишка з’єднується напряму з прямою кишкою та ансусом.
При подібних операціях ускладнення трапляються досить рідко. Діти, яких було прооперовано, можуть контролювати дефекацію, в них відновлюється нормальна перистальтика. Надалі вони ведуть цілком нормальне життя.
Профілактика:
Хворобу Гіршпрунга неможливо попередити.
Переглянуто редакційною колегією I.B.I.S.: 11/06/2002
Хвороба Гірке
(Von Gierke Disease)
Тетяна Дмитрівна Загорулько
Лікар-неонатолог
Волинського обласного дитячого територіального медичного об’єднання
Загальний огляд глікогенозів:
Основні запаси глікогену знаходяться в печінці і скелетних м’язах. Глікоген печінки і м’язів використовується в залежності від потреби організму (лабільний глікоген). Глікоген нервових клітин, провідної системи серця, ендотелію, епітеліальних покривів, слизової оболонки матки, сполучної тканини, ембріональних тканин, хрящів, лейкоцитів є необхідним компонентом клітин, і його вміст не піддається помітним коливанням (стабільний глікоген). Регуляція обміну вуглеводнів здійснюється нейроендокринним шляхом: гіпоталамус, гіпофіз (АКТГ, тиреотропний, соматотропний гормони), β-клітини підшлункової залози (інсулін), наднирники (глюкокортикоїди, адреналін) і щитовидної залози. Порушення вмісту глікогену проявляються в зменшенні або збільшенні кількості його в тканинах і появі там, де він, як правило, не виявляється. Ці порушення найбільш виражені при цукровому діабеті і при спадкових вуглеводних дистрофіях – глікогенозах. Останні зумовлені відсутністю або недостатністю фермента, що приймає участь в розщепленні депонованого глікогену, і тому належать до спадкових ферментопатій, або хвороб накопичення. На даний час добре вивчені 6 типів глікогенозів, зумовлених спадковою недостатністю шести різноманітних ферментів. Це хвороби Гірке (І тип), Помпе (II тип), Мак-Ардля (V тип) і Герса (VI тип), при яких структура накопиченого в тканинах глікогену не порушена, і хвороби Форбса-Корі (III тип), і Андерсена (IV тип), при яких вона різко змінена.
Глікогенози
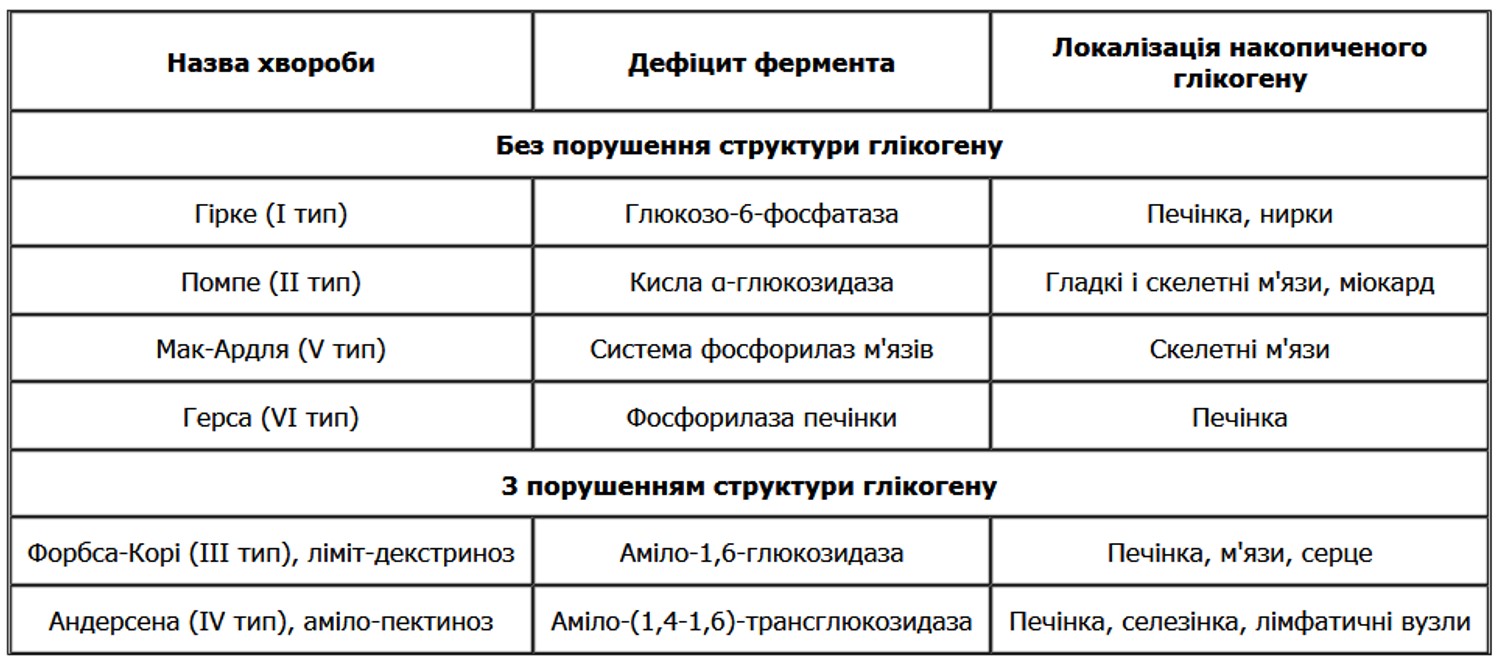
Визначення:
Хвороба Гірке (синоніми: недостатність глюкозо-6-фосфатази, глікогеноз типу І, глікогенна гепатонефромегалія) – спадкова ферментопатія, зумовлена відсутністю фермента глюкозо-6-фосфатази, що приймає участь в розщепленні депонованого глікогену, наслідком цього дефіциту є накопичення глікогену в печінці і нирках. Синдром вперше був описаний Гірке у 1929 році, як поєднання збільшеної печінки і порушення метаболізму глікогену. Захворювання зумовлене дефектами генетичного матеріалу.
Діагностичні критерії:
Відставання в зрості, гепатомегалія, гіпоглікемія, гіперлактатацидемія.
Частота:
Глікогеноз І типу зустрічається з частотою 1 випадок на 100 тис. новонароджених. Питома вага хвороби Гірке серед усіх глікогенозів становить 25 відсотків. Однак, клінічний перебіг саме глікогенозу І типу вважається найважчим.
Тип успадкування:
Аутосомно-рецесивний.
Етіологія:
Глікогеноз І типу спричинений дефектом глюкозо-6-фосфатази, яка каталізує розпад глюкозо-6-фосфату на фосфат і глюкозу. Ця реакція відіграє ключову роль в регуляції глікемії, оскільки вона контролює кінцевий крок в процесах глікогенолізу і глюкогенезу. В нормі глюкозо-6-фосфат, який утворюється в процесі метаболізму глікогену, в печінці під дією глюкозо-6-фосфатази перетворюється у вільну глюкозу, яка надходить у кров і використовується в інших органах; в м’язах, які не містять глюкозо-6-фосфатази, глюкозо-6-фосфат використовується для власних енергетичних потреб, окислюючись аеробним або анаеробним шляхом до лактату.
В 1952 році Корі довів, що хвороба спричинена дефіцитом глюкозо-6-фосфатази. Пізніше дослідження показали, що у деяких пацієнтів не спостерігається дефіциту глюкозо-6-фосфатази, хоча багаточисельні функціональні тести демонструють неспроможність гідролізувати глюкозо-6-фосфат in vivo. Цей стан був названий глікогенозом типу І. Припускається, що він зумовлений дефіцитом глюкозо-6-фосфатспецифічної транслокази, яке забезпечує вхід глюкозо-6-фосфатази (G6Pase) в пластинки ендоплазматичного ретикулуму.
Отже, недостатність глюкозо-6-фосфатази (G6Pase) спричинює глікогеноз типу Іa, a дефіцит глюкозо-6-фосфаттранслокази (G6PT1) – глікогеноз типу Іb. Першу гіпотезу про механізм виникнення глікогенозу типу Іb сформулював Arion. Nordlie описав пацієнта з глікогенозом типу Іс, з дефіцитом транслокази, специфічної до фосфату (G6PT2), яка сприяє проникненню фосфату за межі ендоплазматичного ретикулуму в цитоплазму, завершуючи таким чином гідроліз глюкозо-6-фосфату.
Існування глікогенозу типу Іd, властивого людям з дефіцитом транслокази (G6PT3), здатної виводити глюкозу з ендоплазматичного ретикулуму, ніколи не було доведене достовірно. Ген, відповідальний за G6Pase, локалізований на хромосомі 17q21; за глюкозо-6-фосфат-транслоказу – на 11 хромосомі (11q23).
Патофізіологія:
Печінка не здатна виконувати свою функцію як глюкозо-гомеостатичного органу. Швидке падіння рівня глюкози в крові, що в нормі спричиняється в перші декілька годин після народження (рефлекторне споживання материнської глюкози), не може компенсуватись звичайними гомеостатичними механізмами, таким чином дефіцит глюкози зростає. Наслідком тяжкої гіпоглікемії (іноді глюкоза взагалі не визначається в крові) можуть бути судоми, апное, ціаноз, ушкодження ЦНС. Дефіцит глюкозо-6-фосфатази зумовлює утворення з глюкозо-6-фосфату лактату. Лактатемія прямо пропорційна ступеню стимуляції розпаду глікогену. Акумуляція молочної кислоти в крові спричиняє ацидоз з великою аніонною різницею.
Anion gap = [Na+] – ([Сl] + [НСО3]) = 12 ± 4 мекв/л
В нормі концентрація аніонів в крові дорівнює концентрації катіонів, зменшення в плазмі бікарбонатів збалансовано підвищенням концентрації інших аніонів. Збільшення величини “anion gap” свідчить про збільшення в крові невимірюваних аніонів, тобто про ацидоз. Величезне збільшення фосфорильованих проміжних компонентів гліколізу конкурентно пригнічує рефосфориляцію аденінових нуклеотидів, сприяє деградації нуклеїнових кислот, наслідком чого є збільшення в крові сечової кислоти. Гіперурикемія може досягати рівня, що вимагає застосування інгібіторів ксантиноксидази для попередження нефролітіазу. Тяжка гіпоглікемія стимулює секрецію гормонів мозкової речовини наднирників, які активують ліпопротеїнліпазу і звільняють кількість жирних кислот. Останні в свою чергу транспортуються до печінки, де використовуються для синтезу тригліцеридів і виводяться з печінки як ліпопротеїди низької густини. Навіть при значній гіпоглікемії у пацієнтів з І типом глікогенозу не розвивається значний кетоз, оскільки надлишок ацетил-коензиму А (СоА), який виникає внаслідок гліколізу, активує ацетил СоАкарбоксилазу, що стимулює утворення малоніл СоА на першому етапі синтезу жирних кислот. Малоніл пригнічує транспорт жирних кислот в мітохондріях, тому їх β-окислення і виділення енергії для підтримки клітин не виникає. Ці причини поглиблюють зменшення рівня глюкози і пояснюють відсутність кетонових тіл.
Припускають, що причиною носових кровотеч є пошкодження синтезу мембранних глікопротеїдів, хоча це не дає вичерпного пояснення щодо дефекту агрегації тромбоцитів.
При глікогенозу типу Іb пацієнти є сприйнятливі до грам-позитивної флори. Для нейтрофілів у хворих глікогенозом типу Іb характерний послаблений кисневий вибух на інфекційний стимул. Це зумовлює нейтропенію та зниження індивідуальної стійкості до інфекцій. При кисневому вибусі утворюється супероксид – головний засіб захисту від грам-позитивної флори.
Клініка:
Діти відразу після народження виглядають здоровими, та хвороба маніфестує вже з перших днів життя у вигляді гепатомегалії. Печінка протягом першого року життя м’яка при пальпації, потім стає щільною, вузлуватою. Зовнішній вигляд хворого характерний: маленький зріст – постійна ознака хвороби Гірке, повні щічки, кругле лялькоподібне обличчя, ксантоми (зумовлені підшкірним відкладанням жиру), виступаючий живіт, що різко контрастує з тонкими кінцівками. На шкірі можна побачити численні екхімози, пацієнт скаржиться на часті носові кровотечі, що є наслідком дисфункції тромбоцитів. М’язи атрофовані. Можуть бути присутні ознаки подагри (з боку суглобів, нирок). Хворі відстають у статевому дозріванні, хоча фертильність не порушується, у жінок, хворих глікогенозом І типу, описані успішні вагітності, які були завершені кесарським розтином на 35 тижні гестаційного розвитку і народженням здорових дітей. Хворі не адаптовані до голодування, внаслідок гіпоглікемії можуть виникнути судоми. Внаслідок відкладення глікогену в нирках, останні також збільшуються в розмірах.
Виникнення ускладнень можна затримати за допомогою контролю за метаболічними процесами. Серед ускладнень виділяють: печінкова аденома з ризиком малігнізації, анемія, остеопенія (переломи), кисти яйників.
Маніфестація ниркових ускладнень розпочинається з прихованої клубочкової гіперфільтрації, яка передує розвитку протеїнурії, що може перерости в ниркову недостатність. Поширені гіперкальційурія, нефрокальциноз і камені в сечовивідних шляхах.
Пошкоджена функція нирок у деяких хворих є причиною виникнення артеріальної гіпертензії.
Вважається, що дисліпідемія може стати причиною виникнення панкреатиту, атеросклерозу та серцево-судинних ускладнень. Легенева гіпертензія – рідкісне ускладнення і прогноз його виникнення надзвичайно несприятливий.
Глікогеноз типу Іb має подібну клінічну картину. Крім ознак, характерних для типу Іа, типу Іb властиві нейтропенія, порушення функції моноцитів, що сприяє розвитку частих вторинних інфекційних захворювань, виразковуванню слизових кишківника та ротової порожнини, розвитку запальних кишкових захворювань.
Діагностика:
- Загальний аналіз крові: анемія, нейтропенія.
- Першочергові лабораторні дослідження мають включати визначення глюкози в крові, електроліти.
- Дослідження функцій печінки і нирок; кількість сечової кислоти в крові, креатиніну, кліренс сечовини, креатиніну, рівень білірубіну та трансаміназ.
- Коагулограма – зміни характерні для порушення функцій тромбоцитів (кількість тромбоцитів збільшена), зменшення фібриногену та інших факторів зсідання крові.
- УЗД нирок, печінки.
- Морфологічна діагностика того чи іншого типу можлива при біопсії з допомогою гістоферментативних методів.
- Інші методи:
- проба з глюкагоном (1 мг/м² поверхні тіла): гіпоглікемія не зменшується, проте збільшується кількість лактату.
- проба з галактозою (1,75 г/кг) не спричиняє змін глюкози в крові, хоча зростає кількість молочної кислоти.
Лікування:
В лікуванні важливе місце посідає дієта, скерована на уникнення метаболічних розладів, запобігання ускладненням з боку нирок і печінки, досягнення зросту згідно вікової норми. Лікування складається з частого годування, безперервного нічного ентерального живлення, вживання вуглеводів, що повільно всмоктуються і обмеження вживання фруктози і галактози, які можуть посилювати гіперлактатемію. Калорійність добового раціону має бути суворо контрольована. Недостатнє харчування не відкорегує метаболічні порушення (гіпоглікемію, гіперлактатемію, гіперурикемію); надлишкове харчування збільшує накопичення глікогену, гепатомегалію, гіперліпідемію, спричинює ожиріння. Дієта має містити велику кількість важкозасвоюваних вуглеводів (60-65% ккал від добової калорійності) і малу кількість ліпідів (20-25% від добової калорійності).
Для новонароджених і малят корисні часті годування і введення безперервно через назогастральний зонд їжі, забезпечуючи спочатку 8-10 мг глюкози/кг/хв, потім 5-7 мг/кг/хв.
Додаткова терапія включає вітаміни, кальцій для попередження остеопенії і аллопуринол, коли присутня гіперурикемія.
Для попередження ускладнень з боку нирок ефективним є застосування інгібіторів ксантинонидази. Для корегування анемії застосовують препарати заліза.
Для вагітних цей препарат безпечний.
Корегувати нейтропенію при глікогенозі тип Іb може гранулоцитарно-макрофагальний колонієстимулюючий фактор, що зменшує тяжкість бактеріальних інфекцій і полегшує перебіг запальних кишкових явищ.
Filgrastim (G-CSF, Neupogen) активує, стимулює продукцію, дозрівання, міграцію і цитотоксичність нейтрофілів. Доза 5 мкг/кг для дітей та дорослих. Протипокази: гіперчутливість до препарату. Взаємодія: синергічний ефект з ІЛ-3 (збільшення кількості мегакаріоцитів і тромбоцитів). Вагітність: безпечність для вагітних не доведена. Застереження: не застосовувати 12-24 год. після цитотоксичної хіміотерапії, оскільки підвищується чутливість мітотичних мієлоїдних клітин до хіміотерапії; не вживати з содою; застосовувати обережно при подагрі, псоріазі. Побічні ефекти: гарячка, біль в кістках, грипоподібні симптоми.
Ефективність лікування оцінюється за допомогою моніторування таких показників – крива росту, ступінь гепатомегалії, артеріальний тиск, біохімічних параметрів – рівень глюкози в крові більше 3,5 ммоль/л, лактат сечі менше 0,6 ммоль/л в нічній і денній сечі, тригліцеридемія, холестеринемія, урикемія, протеїнурія і загальний аналіз крові.
Лікування ускладнень полягає у трансплантації печінки після безуспішної терапії дієтою. Трансплантація печінки корегує гіпоглікемію та інші біохімічні показники, але не нормалізує кількість нейтрофілів, також не доведено, що це може попередити ураження нирок. Трансплантація нирок, проведена у випадках тяжкої ниркової недостатності, не корегує гіпоглікемії. Можливість одночасної пересадки печінки і нирок обговорюється, але такі операції не проводились. Під час хірургічних втручань, з огляду на ризик виникнення кровотеч і метаболічного дизбалансу необхідний постійний моніторинг глікемії, лактацидемії, системи згортання крові. Глікемія має підтримуватись перфузіями 10% глюкози перед, протягом і під час оперативного втручання. Заборонено застосовувати розчини, що містять лактат (наприклад розчин Рінгера). Пацієнти, які пройшли терапію, і у яких було досягнуте клінічне і лабораторне видужання, підлягають диспансерному нагляду і повинні проходити повний медичний огляд кожні 6 місяців.
Акушерська тактика:
Небезпека виникнення кровотеч і гіпоглікемії потребують постійного моніторування вагітностей для запобігання мимовільним викидням і мертвонародженням. Рекомендовано розродження шляхом кесарського розтину приблизно на 35 тижні вагітності.
Диференційний діагноз:
Інші типи глікогенозів, гепатомегалія іншої етіології.
Прогноз:
Залежить від дотримання дієти, запобігання виникненню ускладнень.
Номер з каталогу МІМ:
232200 Glycogen Storage Disease I.
Література:
- Deeksha Bali, Yuan-Tsong Chen, Stephanie Austin, Jennifer Goldstein. Glycogen Storage Disease Type I (https://www.ncbi.nlm.nih.gov/books/NBK1312/).
- Jones KL. Smith’s Recognizable Patterns of Human Malformation. Philadelphia: W.B.Saunders Company 1997:680.
Переглянуто редакційною колегією I.B.I.S.: 16/08/2004
|
|
|
|
|
|






{kind=link}