
Serhiy
Екстрофія сечового міхура
(Bladder Exstrophy)
І.С. Кулик
Студентка Хмельницького Технологічного Університету Поділля
Що таке екстрофія сечового міхура?
Слово “екстрофія” походить від грецького ekstrophien, що означає “навиворіт”. Екстрофія сечового міхура – це вада, при якій міхур та структури, що до нього прилягають, вивернуті назовні. Передня черевна стінка не покриває ці органи.
Які інші аномалії пов’язані з екстрофією сечового міхура?
Дитина з таким діагнозом, зазвичай, має усі, або деякі з перелічених нижче аномалій:
- Епіспадія – у хлопчиків уретра (канал, через який випорожнюється сечовий міхур) повністю не сформована і може бути надзвичайно короткою і роз’єднаною. Саме через це отвір уретри знаходиться не на кінці пеніса, а на верхній його поверхні. Якщо відсутнє змикання уретри на всьому протязі статевого члена, то він буде плоским та коротким, по формі нагадуватиме маленьку лопатку. У дівчаток отвір сечовивідного каналу розташований між клітором та малими статевими губами.
- Дорсальна хорда – викривлення статевого члена внаслідок диспропорційного розвитку сухожиль (у верхній частині пеніса сухожилля коротші).
- Відсутність шийки сечового міхура та сфінктера. Шийка сечового міхура – це канал, розташований у нижній частині міхура. В цьому каналі знаходиться кільце м’язів, які відкривають і закривають отвір міхура. Саме це м’язеве кільце (сфінктер) дозволяє утримувати сечу. Якщо сфінктер відсутній, то контроль сечовиділення неможливий.
- Малий об’єм сечового міхура. Міхур не в змозі утримувати нормальну кількість сечі.
- Аномальне розташування сечоводів – каналів, по яких сеча потрапляє з нирок до сечового міхура. Порушене з’єднання сечоводів із сечовим міхуром спричиняє відтік сечі у зворотньому напрямку, так званий рефлюкс.
- Діастаз лобкових кісток – великі кістки, що утворюють пояс нижніх кінцівок, зазвичай формують коло. Таз підтримує сечовий міхур, статеві органи і органи травлення. З’єднані лобкові кістки формують передню частину тазу та захищають сечовий міхур, уретру та м’язи черевної стінки. При екстрофії сечового міхура, лобкові кістки не з’єднуються, утворюючи широку порожнину. Причиною такого відкритого тазового кола може бути вивих стегон. Це змушує дитину з екстрофією ставити ноги носками урізнобіч, коли вона стоїть чи іде. Однак, діти з екстрофією сечового міхура можуть гратись, бігати, стрибати, не дивлячись на аномалію тазу.
- Анус розміщений нижче, ніж звичайно. Отвір прямої кишки розташований набагато нижче (ближче до калитки або піхви). Як правило, таке розташування не супроводжується порушенням функції прямої кишки.
- Низьке розташування пупка. Пупок не завжди можна помітити і він може бути зміщеним, коли сечовий міхур був закритий хірургічний шляхом.
Екстрофія сечового міхура у хлопчиків:
У хлопчиків може бути короткий, викривлений пеніс, дещо сплощений на верхівці. Характерною є епіспадія. Спостерігаються також двобічні пахові кили, крипторхізм. Хірургічне втручання потрібне буде для пластики пенісу та відновлення його функцій.
Екстрофія сечового міхура у дівчаток:
У дівчаток з екстрофією сечового міхура внутрішні статеві органи (матка, маткові труби та яйники) розвинені правильно. Піхва може бути дещо звуженою та розміщуватись вище, ніж звичайно. Клітор – роздвоєний, не з’єднані статеві губи, що хірургічно корегується.
Дорослі пацієнтки з екстрофією сечового міхура можуть вести нормальне статеве життя. Більшість із них матиме здорових дітей. Але перед плануванням вагітності, необхідно проконсультуватись у лікаря, щоб запобігти пролапсу матки, спланувати тактику ведення.
Коли виникає екстрофія?
Фактори, які викликають екстрофію, діють на ранніх стадіях ембріонального розвитку – 4-5 тижнів після запліднення. Причини та механізми розвитку цієї хвороби остаточно не досліджені. Існує декілька точок зору на шляхи формування екстрофії сечового міхура: згідно з однією, вада виникає внаслідок порушення процесу змикання (з’єднання) клоакальної мембрани. Інша група вчених припускає, що відбувається розрив тонкого шару клітин, що вкривають зародковий сечовий міхур, і останній вивертається назовні. У будь-якому разі, для сучасної медичної науки залишається актуальним питання з’ясування причин виникнення екстрофії сечового міхура.
Що визначає ступінь екстрофії?
Ступінь екстрофії залежить від розміру отвору. Екстрофію можна уявити як ряд вад, які варіюються у складності – від епіспадії (вада уретри та отвору) до найскладніших – клоакальної екстрофії (дефект уретри, сечового міхура та кишківника) та класичної екстрофії сечового міхура (дефект сечового міхура та уретри).
Як часто трапляється екстрофія сечового міхура?
Хоча екстрофія виявляється у 1 на 30000 новонароджених, вона може передаватись спадково. За результатами деяких досліджень, існує 1 шанс із 100, що інші брати та сестри дитини з екстрофією сечового міхура матимуть цю ваду та 1 із 70, що діти пацієнта з екстрофією успадкують її. До того ж, екстрофія сечового міхура у 2-3 рази частіше зустрічається у чоловіків, ніж у жінок.
Як лікують хворих з екстрофією сечового міхура та поєднані з нею аномалії?
Екстрофія сечового міхура вимагає хірургічного лікування. Курс лікування залежатиме від типу та ступеня вираженості аномалії. Як не існує двох дітей з однаковими екстрофіями, так і немає однакових курсів лікування при цій ваді. Постійні та тривалі пошуки шляхів удосконалення хірургічних методів лікування екстрофії сечового міхура суттєво покращили перспективи для пацієнтів щодо якості та тривалості життя.
Як довго триватиме лікування?
На сьогодні лікування вимагає серії хірургічних втручань, які проводять протягом кількох років. Такий курс лікування називається поступовим відновленням. Точний час проведення, тип та результати хірургічного втручання залежатимуть від складності та ступеня вираженості вади.
Які цілі лікування?
Цілями поступового відновлення є:
- закриття сечового міхура та уретри;
- закриття черевної стінки;
- збереження функції нирок та репродуктивної здатності;
- задовільний зовнішній вигляд чоловічих та жіночих статевих органів;
- психологічно врівноважена дитина та родина.
Як зазвичай призначається лікування?
Нижче подано огляд поступового відновлення:
- первинне закриття сечового міхура проводиться у перші 24-48 годин після народження дитини. Під час цієї операції розщеплені лобкові кістки з’єднуються, сечовий міхур закривається, розміщується у таз та закривається черевна стінка. Проводиться спеціальна пластика, щоб сечовий міхур міг утримувати сечу;
- відновлення геніталій хлопчиків та дівчаток, як правило, проводиться у різний час. У дівчаток відновлення геніталій відбувається під час першої операції. У хлопчиків подібне хірургічне втручання проводиться між першим та другим роком життя. Воно має тут велике значення, оскільки, результатом операції є уретральний опір, сеча накопичується в сечовому міхурі, що призводить до збільшення його об’єму;
- відновлення шийки сечового міхура проводиться для того, щоб дитина могла самостійно контролювати сечовиділення. Така операція проводиться у віці 5 років, до початку навчання в школі. Під час цієї операції сечовід переміщують так, щоб запобігти рефлюксу сечі назад у нирки, а сфінктер міг відкриватись та закриватись тільки тоді, коли сечовий міхур повний.
Переглянуто редакційною колегією I.B.I.S.: 09/04/2003
Екстрофія клоаки
(Cloacal Exstrophy)
Д.Р. Ахмеджанова
Студентка Хмельницького Технологічного Університету Поділля
Екстрофія клоаки (ЕК) – це рідкісна і складна вроджена вада, що виявляється у одного із 250000 новонароджених. Екстрофія клоаки часто має різні назви, а саме – міхурово-кишкова щілина, екстрофія клоаки, комплекс OEIS.
Опис комплексу ОЕIS:
О – omphalocele (омфалоцеле) – це вада передньої черевної стінки, при якій внутрішні органи, вкриті тонкою мембраною, знаходяться поза черевною порожниною.
Е – exstrophy (екстрофія клоаки) – сечовий міхур відкритий, а пряма кишка відкривається у нього.
I – impеrforate anus (неперфорований анус) – товста кишка сполучена з сечовим міхуром.
S – spina bifida (спинномозкові вади) – як важкі, так і малі аномалії. У багатьох випадках діти з екстрофією клоаки народжуються зі Spina bifida.
Важкість вад при народженні визначатиме план дій щодо подальшого життя дитини. Потрібно мати на увазі, що кожний окремий випадок вимагатиме особливого підходу, залежно від того, чи вади будуть множинними чи ізольованими.
Який прогноз стосовно життя та здоров’я?
З огляду на розвиток сучасної медицини, дитина з ЕК має усі шанси вижити. Однак, якість життя залежатиме від складності вроджених вад. П’ятнадцять років тому багато дітей з подібними вадами помирали при народженні або в перші дні життя. Десять років тому діти з такими вадами мали хороші прогнози щодо життя та розвитку. П’ять років тому, з розвитком медичної науки ці діти отримали майбутнє, сповнене надій, що вони стануть повноцінними членами суспільства.
Що є причиною таких вад?
Причина екстрофії клоаки остаточно нез’ясована. Однак, ваду можна діагностувати пренатально.
Чи постраждають розумові здібності дитини з ЕК?
Більшість дітей з екстрофією клоаки дуже розумні і немає підтвердження того, що вони мають нижчий рівень інтелекту, ніж усі інші.
Проблеми з визначенням статі:
Якщо новонароджена дитина чоловічої статі, то хірурги порадять змінити стать на жіночу. Це виконується шляхом видалення яєчок та формування піхви для можливості статевого життя у подальшому.
Які операції потрібні дитині з ЕК?
Протягом життя дитина потребуватиме ряду операцій. Іноді, під час однієї операції вдається досягти декількох цілей.
Яким буде перше оперативне втручання?
Через деякий час після народження дитині проведуть першу операцію по усуненню кили. Хірурги створять прохід для виведення калу, а також закриють сечовий міхур. В цей же час можна виправити і спинномозкові вади. На закінчення операції можливе накладання стоми – штучного сполучення для виведення сечі та калу.
Чи залишатиметься цей пристрій назавжди?
Незалежно від того, чи проводиться операція низведення з метою формування прямої кишки чи закриття стоми, визначальним все ж є величина прямої кишки новонародженої дитини. Якщо дитина народилась з достатньою величиною товстої кишки, то проводиться операція повного низведення. Навіть якщо дитина не може утримувати кал, організм очишається за допомогою щоденних клізм. Якщо ж у новонародженого недостатньо сформована пряма кишка, то необхідною буде стома протягом усього життя.
Потреба у пристрої для виведення сечі, можливо, з часом, відпаде.
А що ж далі?
Наступним кроком є оздоровлення дитини, підготовка її до життя в домашніх умовах. Це означає, що вона отримуватиме належний післяопераційний догляд. Також робитиметься все можливе, щоб дитина набрала відповідну її віку масу, якщо вона народилась передчасно. В цей час батьки зіштовхуються з рядом фізіологічних та емоційних потреб дитини.
Що буде після повернення до дому?
Перші дні – найважчі. Батьки і дитина звикатимуть до цього, і, можливо, їм буде необхідна допомога медсестри. Через деякий час все налагодиться. Наступні 3-4 роки життя будуть найлегшими. Вроджені вади дитини не будуть помітними для усіх. Більшість людей не знатиме про це, якщо батьки не скажуть. Можуть виникнути лише невеличкі проблеми, наприклад – чутливість до зневоднення або подразнення від пристроїв для виведення сечі. Зазвичай, протягом цих років виникає найменше проблем.
Які наструпні операції?
Найголовніша операція для відновлення виведення сечі проводиться для того, щоб дитина могла самостійно випорожнюватись. При цьому формується новий сечовий міхур з клапанним механізмом для періодичної катетеризації. У цей же час можна сформувати піхву.
Як щодо можливості статевого життя?
Якщо ваша дитина жіночої статі, то вона зможе жити статевим життям. Якщо ж – чоловічої, то потрібна буде хірургічна зміна статі з формуванням нео-піхви.
Чи можуть пацієнти з ЕК мати дітей?
У багатьох випадках відповідь на це запитання – позитивна. Це залежатиме від того, чи є яєчники та матка. Якщо ж так, то дуже ймовірно, що пацієнт з ЕК зможе мати дітей. Фактично, з деяких причин, вони дуже легко вагітніють.
Зрозуміло, життя родини, де народилась дитина з ЕК зміниться і, можливо, буде складнішим, ніж у інших. Після народження дитини доведеться відповісти на питання друзів та сім’ї. Коли дитина досягне шкільного віку, у людей навколо теж виникнуть певні питання. Прогрес у медицині відкриватиме нові обрії для пацієнтів з ЕК.
Переглянуто редакційною колегією I.B.I.S.: 07/04/2003
Діагностика, лікування та профілактика муковісцидозу
(Mucoviscidosis, Cystic Fibrosis)
Методичні рекомендації
М.Л. Аряєв, Н.А. Кононенко, О.О. Старець,
Ю.Г. Циунчик, О.Г. Шаповалов, О.Г. Ліман,
Л.Л. Поплавська, Л.П. Михайлець, Н.Г. Горовенко
Одеський державний медичний університет,
Інститут педіатрії, акушерства та гінекології АМН України,
Київська медична академія післядипломної освіти ім. П.Л. Шупика
Вступ:
Методичні рекомендації відображують сучасні підходи до діагностики та лікування муковісцидозу, що сформувалися після видання попередніх рекомендацій МОЗ УРСР “Этапное лечение муковисцидоза”, Одесса, 1987. Важливе місце відведено особливостям клінічної картини, методам параклінічної діагностики. Викладені принципи евакуації бронхіального секрету та боротьби з інфекцією дихальних шляхів, питання дієти та ферментотерапії з точки зору стратегії безперервного спостереження за хворими. Висвітлені проблеми медико-генетичного консультування.
Методичні рекомендації призначені для педіатрів, генетиків, пульмонологів, гастроентерологів, студентів медичних університетів, лікарів-інтернів, магістрів, клінічних ординаторів.
Муковісцидоз (МВ) – це спадкове аутосомно-рецесивне мультисистемне захворювання, що розвивається на підставі продукції екзокринними залозами секрету підвищенної в’язкості з розвитком вторинних змін переважно у органах систем дихання та травлення.
Епідеміологія МВ:
МВ – одне з найчастіших спадкових захворювань. Нині відомо, що МВ виявляється серед представників усіх рас і національностей, хоч переважно розповсюджений серед європеоїдів з частотою у середньому 1 випадок на 2500-3000 новонароджених (Росія – 1:3800; Велибританія – 1:2500, Україна – 1:2300 новонароджених). Щорічно у світі народжується більш ніж 45000 хворих, у Америці – 1000 , Велибританії, Франції – 500-700, Росії – 300.
Середня тривалість життя хворих на МВ у країнах Західної Європи, Австралії складає 26-30 років. В Україні цей показник з 2-3 років (1970-1980 р.р.) досягнув 12-14 років (1995-1998 р.р.).
Етіологія і патогенез МВ:
В 1985 році стало можливим локалізувати ген МВ у довгому плечі 7 хромосоми, а потім у 1989 році ізолювати його і розшифрувати структуру. Ген МВ вміщує 27 екзонів і охоплює 250 тисяч пар основ. Він визначає вироблення первинного білкового продукту – трансмембранного регуляторного білка муковісцидозу (ТРБМ), який укорінюється в мембрану клітини і є її хлоридним каналом.
Наслідок мутації гена МВ – порушення структури і функцій ТРБМ, що, у свою чергу, веде до порушення транспорту іонів хлору через апікальну частину клітин секреторного епітелію. Результатом цього є збільшення реабсорбції натрію клітинами, зміни електролітного складу і дегідратація секрету екзокринних залоз. Згущення секрету призводить до порушень його евакуації та розвитку вторинних змін з ураженням бронхолегеневої системи, підшлункової залози, травного каналу, печінки, статевих залоз та інших органів.
За даними Консорціуму з вивчення МВ у гені ТРБМ на цей час виявлено більше 720 можливих мутацій, які відносяться до різних типів. Основною мутацією, з якою пов’язана більшість випадків МВ, є делеція F508. Вона визначається як найчастіша і на Україні, де її частота за останніми даними становить 50%. Досить часто зустрічаються мутації N1303К (1,5%) та СFТRdеl21кb (3,5%), мутації R334W, R553Х, G551D і 1677dеlТА зустрічаються лише у поодиноких випадках з частотою менш 1% кожна. В Україні відсоток визначення видів мутацій не перевищує 55%.
Прогрес у вивченні молекулярно-генетичних основ МВ дозволяє з’ясувати значний фенотипічний поліморфізм проявів та перебігу цього спадкового захворювання.
Клінічна характеристика та діагностика муковісцидозу:
Виділяють такі типові форми захворювання:
- змішану (80-85% хворих);
- переважно легеневу (10-15%);
- переважно кишкову (5%).
Атипові форми зустрічаються досить рідко, до них відносять ізольовану електролітну (псевдосиндром Бартера), печінкову та інші з одиничними клінічними проявами.
Бронхолегеневі зміни у 80-95% випадків домінують у клінічній картині, визначаючи прогноз у більшості хворих на МВ. Основним симптомом маніфестації ураження дихальної системи у дітей є кашель, що може виникати вже у перші тижні життя. Спочатку він непродуктивний, проявляється у вигляді частих покашлювань, потім стає нападоподібним, з мокротинням, що важко виділяється, з часом набуває хронічного характеру. Поступово з’являється задишка. Порушення мукоціліарного транспорту у дихальних шляхах призводить до застою бронхіального секрету, створює умови до раннього вторгнення патогенної мікрофлори та розвитку запального процесу з гіперсекрецією мокротиння. Таким чином, формується замкнене коло.
Поступово мукостаз, бронхіальна обструкція та хронічний запальний процес призводять до розвитку прогресуючих незворотніх змін, що характеризуються наявністю бронхоектазів, пневмосклерозу, зон обмеженого пневмофіброзу, емфіземи легенів. Різко знижуються функціональні можливості легенів, виражені симптоми хронічної гіпоксії, з’являються ознаки “легеневого серця” та інші ускладнення, формується типовий вигляд таких хворих: відставання у фізичному розвитку, деформована грудна клітина, збільшена у передньо – задньому розмірі, худі кінцівки з деформацією дистальних фаланг пальців у вигляді “барабанних паличок”, суха, сірувато-землиста шкіра, ціаноз, виражена задишка, мученицький кашель з в’язким гнійним мокротинням.
Про загострення свідчить поява слідуючих симптомів та ознак: збільшення частоти чи тривалості кашлю, зміна кількості або кольору мокротиння, посилення задишки, тахіпное, участь додаткових м’язів у диханні, поява кровохаркання, зменшення толерантності до фізичного навантаження, погіршення апетиту, відсутність або негативна динаміка маси тіла, підвищення температури тіла, зниження функції легенів обструктивного типу, нові аускультативні та рентгенологічні зміни, гематологічні ознаки.
Досить рідко, переважно у дітей першого року життя, спостерігається тривалий або рецидивуючий перебіг першої пневмонії навіть при своєчасно розпочатому і адекватно проведеному лікуванні. У хворих пневмонії можуть супроводжуватися утворенням ателектазів, абсцедуванням. Надалі бронхолегеневий процес набуває хронічного перебігу з тенденцією до неухильного прогресування.
Іноді захворювання протягом тривалого часу перебігає безсимптомно, фізичний розвиток дітей майже не порушений, процес у легенях проходить з нечастими загостреннями, досить стабільною рентгенологічною картиною. При цьому частіше спостерігається картина повільно прогресуючого хронічного бронхіту.
Особливістю мікрофлори бронхіального секрету хворих на МВ є переважання двох мікробних агентів: Staphylococcus aureus і Pseudomonas aeruginosa, досить часто зустрічається також Haemophilus influenzae. У ранньому віці звичайно домінує St. aureus, а подалі переважаючим збудником стає Ps. aeruginosa, яка виділяється за даними авторів у 85% хворих після 10-річного віку. Взаємодія Ps. Aeruginosa з організмом при МВ відбувається у дві фази:
- ініціальна інфекція з непостійною ізоляцією мікроорганізмів, що асоційована iз звичайними штамами;
- хронічна ендобронхіальна колонізація з персистенцією інфекції.
Саме ця фаза сприяє прогресуванню хронічного запального процесу у бронхолегеневій системі з розвитком вторинних імунних змін. Вона пов’язана з конверсією звичайних штамів у мукоїдні, що здатні синтезувати слизоподібну оболонку, яка підвищує в’язкість мокротиння, перешкоджує ерадикації мікроорганизму. Вкрай несприятливим є колонізація майже антибіотикорезистентним мікроорганізмом – Burkholderia cepacia.
Бронхолегеневий процес при МВ може дати ряд ускладнень: легеневу кровотечу (5%), пневмоторакс (2-5%), піопневмоторакс (3%). До ускладнень відносять також розвиток алергічного бронхопульмонального аспергільозу (АБПА) внаслідок сенсибілізації до антигенів Aspergillus fumigatus. Характерні ознаки – бронхоспазм, еозінофілія у крові та мокротинні, значне зростання рівня IgE, знаходження Aspergillus у бронхіальному секреті, рентгенологічні зміни у вигляді легеневих інфільтратів, сегментарних або дольових ателектазів.
Дослідження функції зовнішнього дихання у більшості хворих виявляють комбінацію обструктивних і рестриктивних порушень. Рестриктивні зміни, особливо при загостренні бронхолегеневого процесу, маскуються обструктивним процесом, головним чином, у дрібних бронхах. Поряд з порушеннями у розподілі газу, що вдихається, при МВ є нерівномірність в розподілі вентиляційно-перфузійних відношень та порушення дифузії в легенях, що призводить до гіперкапнічної гіпоксії.
Рентгенологічна характеристика МВ: ведучими слід вважати зміни інтерстицiальної тканини, розташованої перибронхіально, що виявлюється у посиленні легеневого малюнка, появі сітчатості, тяжистості. Деформацію легеневого малюнка супроводжують ділянки нерівномірного затініння, інфільтративні зміни. Порушення бронхіальної провідності обумовлює рентгенологічні прояви емфіземи легенів у вигляді підвищення прозорості легеневої тканини, викривлення грудини з появою ретростернального простору, ущільнення куполів діафрагми, розширення міжреберних проміжків. У багатьох дітей серце “висить” на судинах, здається крапельним. У деяких хворих фіброз в прикореневих зонах виявлений настільки, що серце ніби “замуровано” у фіброзно зміненій тканині легенів.
Бронхоскопія виявляє картину двобічного ендобронхіту, підвищену в’язкість мокротиння, його скловидний характер, а також симптом нашарування секрету на стінках головних бронхів і трахеї. Характерні гнійні “пробки”, що локалізуються у трахеобронхіальних і бронхіальних кутах.
Важкість перебігу МВ посилюється розвитком вторинних дефектів в імунному статусі хворих. Гуморальний та клітковий імунітет у хворих звичайно не змінені, однак, при прогресуванні легеневої інфекції може визначатися функціональний дефіцит кліткового імунітету. Порушені процеси фагоцитозу, особливо у хворих, які колонізовані синьогнійною паличкою, відзначaється знижений рівень секреторного імуноглобуліну А, визначаються у високому титрі антитіла до синьогнійної палички, циркулюючі імунні комплекси.
Зміни в травній системі при МВ виникають, головним чином, внаслідок панкреатичної недостатності у 85-90% хворих, яка вже протягом перших місяців життя обумовлює розвиток синдрому мальабсорбції. У невеликої частини хворих симптоми недостатньої функції підшлункової залози з’являються пізніше, звичайно на протязі першого року життя. Характерними є часті випорожнення, кількість збільшена, вони світлі за кольором, жирні, замазкоподібні, з неприємним запахом. Про значну стеаторею свідчать поява на пелюшках плям, що важко прасуються, видимі очима капельки жиру. Появою метеоризму, а також гіпотонією м’язів пояснюється збільшений живіт. У чверті хворих з важким перебігом кишкового синдрому спостерігається розвиток вторинної целіакії, дисахаридазної недостатності, ексудативної ентеропатії. Значні порушення усмоктування призводять до гіпопротеінемії з набряками та анемією, значними водно-електролітними змінами. Гіпотрофія при МВ супроводжується ознаками дефіциту жиророзчинних вітамінів: м’язовою гіпотонією, геморагічним синдромом, ламкістю нігтів та волосся, сухістю шкіри, тріщинами у кутах рота та інш.
Маніфестацією МВ у перші дні життя може бути меконіальний ілеус (5 % випадків за даними авторів). До його розвитку призводять панкреатична недостатність, а також порушення секреції слизу на протязі травного каналу, уповільнений пасаж вмісту кишечника з його дегідратацією. Пізніше у частини хворих зустрічається еквівалент меконіального ілеусу – дистальний інтестінальний обструктивний синдром (ДІОС). Для нього характерними є болі у животі, пальпація фекальних мас у правій половині черевної порожнини, симптоми часткової або повної кишкової непрохідності, рентгенологічні зміни у ілеоцекальному куті. Досить рідко ДІОС може бути варіантом маніфестації МВ у дітей шкільного віку. У деяких хворих відмічається схильність до запорів.
Випадіння прямої кишки спостерігається у 1/5 хворих переважно до 5-річного віку. Цей симптом виникає у хворих з панкреатичною недостатністю внаслідок порушень дефекації, а також, зниження тонусу мускулатури промежини.
Приблизно у третини хворих, по більшості старшого віку, як прояв порушеної моторики шлунково-кишкового тракту та впливу інших чинників спостерігається гастро-езофагальний рефлюкс. Він супроводжується нудотою, блювотою, регургітацією їжі, больовим синдромом, дисфагією, погіршується апетит до анорексії. Також він сприяє появі респіраторної симптоматики.
Розлад травлення, недостатнє засвоєння компонентів їжі та підвищені енерговитрати призводять до порушень нутритивного статусу: затримується фізичний розвиток, спостерігаються значний дефіцит довжини та маси тіла, симптоми гіповітамінозу. У більшої частини хворих на муковісцидоз затримується пубертатний розвиток.
У хворих без клінічних ознак панкреатичної недостатності підвищена частота розвитку гострих та хронічних панкреатитів.
Панкреатичну недостатність підтвержують наявністю стеатореї при копрологічному дослідженні (жирові краплі), а також збільшеними добовими втратами жиру з випорожненнями, зменшеним коефіцієнтом абсорбції жиру (КА). Кількість фекального жиру звичайно не перебільшує 5 г на добу за умови достатнього споживання жиру з їжею, нормальний рівень КА жиру дітей до року складає 80-85%, у старших – 90-95%. КА = ((жир їжі – фекальний жир) / жир їжі) х 100%.
Ураження гепатобіліарної системи при МВ обумовлено обструкцією внутрішньопечінкових жовчовивідних шляхів аномально в’язкою жовчю, порушенням метаболізму ендогенних жовчних кислот, значними імунними змінами. Ці чинники призводять до розвитку у 10-15% хворих фокального чи мультилобулярного біліарного цирозу печінки з типовими проявами (гепатомегалія, портальна гіпертензія, спленомегалія, варикозне розширення вен стравоходу, функціональні, ультразвукові, гістологічні зміни). Цироз печінки на протязі тривалого часу не має клінічних симптомів, його частота збільшується з віком хворих. Метаболічні порушення сприяють дифузній жировій дистрофії печінки. Тривала холестатична жовтяниця новонароджених може бути варіантом маніфестації муковісцидозу у 1-2% хворих.
Характерними ознаками муковісцидозу є синусити, ранній носовий поліпоз. Може зустрічатися асимптоматичне збільшення слинних залоз.
Прогресуючий фіброз підшлункової залози веде до ураження її ендокринних елементів зі зниженням толерантності до глюкози, розвитком цукрового діабету у дітей старшого віку та дорослих.
Таким чином, у найчастіших типових випадках характерним є виявлення у дитини перших років життя комбінації ознак ураження системи травлення, затримки фізичного розвитку з респіраторною симптоматикою. Слід пам’ятати, що у хворих на МВ можуть бути інші варіанти маніфестації та проявів захворювання в залежності від віку, які наведено нижче:
Антенатальний період
- При проведенні ультрасонографії як первинні ознаки можуть виявлятися дилатація тонкого кишечника та перитонеальна кальцифікація, що пов’язане з розвитком меконіального ілеусу.
Грудний вік
- Солона на смак шкіра.
- Дуже швидке виникнення зморшок шкіри на пальцях у воді.
- Бронхообструктивний синдром у грудної дитини.
- Гіпотрофія без наявності стеатореї.
- Затяжна обструктивна жовтяниця.
- Псевдосиндром Бартера з гіпонатріємією, гіпокаліємією та метаболічним алкалозом.
- Гемолітична анемія та набряки внаслідок дефіциту вітаміну Е.
- Ознаки дефіциту вітаміну А.
- Геморагічні розлади внаслідок дефіциту вітаміну К.
Ранній вік
- Бронхообструктивний синдром.
- Поліпоз носа.
- Рецидивуючі синусити.
- Тепловий удар та дегідратація в жарку погоду.
- Випадіння прямої кішки.
- Дистальний інтестінальний обструктивний синдром.
Старший вік
- Зниження толерантності до глюкози.
- Збільшення печінки.
- Портальна гіпертензія зі спленомегалією.
- Затримка пубертатного розвитку, малий зріст.
Підлітковий період та дорослі
- Чоловіче непліддя, вроджена двобічна атрезія vas deferens.
- Тепловий удар.
Лабораторна діагностика:
Основним методом лабораторної діагностики залишається потовий тест, який повинен проводитись у всіх випадках підозри на МВ. Стандартний потовий тест з пілокарпіновим електрофорезом у хворих на МВ виявляє підвищення у потовій рідині хлоридів та натрію.
Діагностичним критерієм муковісцидозу є вміст у поті хлоридів та натрію > 60 ммоль/л у дітей і > 70 ммоль/л у підлітків та дорослих за результатами двох або більше тестів, проведених в окремі дні. Сумнівним вважається результат 35-60 ммоль/л, коли потрібне повторне тестування та прискіпливе діагностичне обстеження.
Мінімальна кількість поту, яку можна аналізувати, складає 100 мг.
Ретельно виконаний потовий тест має низький рівень хибнонегативних результатів – менш, ніж 2%, тобто має високу чутливість. Хибнонегативні результати виникають:
- у хворих на МВ з набряками чи гіпопротеїнемією;
- у хворих на МВ, які приймають клоксацилін та його похідні.
Під час аналізу потового тесту слід ураховувати можливість хибнопозитивних результатів при наступних захворюваннях:
- тяжка гіпотрофія та виснаження;
- недостатність надниркових залоз;
- нефрогенний нецукровий діабет;
- нефроз;
- синдром Моріака;
- гіпотиреоїдизм;
- мукополісахаридози;
- ектодермальна дисплазія;
- глікогеноз II типу;
- сімейний холестаз;
- фукозідоз;
- сімейний гіпопаратиреоїдизм;
- ВІЛ-інфекція.
Інтерпретацію результатів слід проводити з урахуванням віку хворого. У здорових новонароджених у перші дні життя рівень електролитів може знаходитись на верхній межі норми чи бути підвищеним. У 5-10% здорових підлітків та дорослих вміст хлоридів та натрію в поті вище, ніж 60 ммоль/л. Усі сумнівні негативні тести повинні перевірятися. Для інтерпретації сумнівних результатів корисні дані, що одержані при паралельному визначенні хлоридів та натрію:
- рівень натрію у поті здорових людей звичайно вище, ніж рівень хлоридів;
- рівень хлоридів у поті хворих на МВ, як правило, вище, ніж рівень натрію;
- сума рівня натрію та хлоридів у здорових дітей звичайно менша ніж 140 ммоль/л, а у хворих на муковісцидоз більша ніж 140 ммоль/л;
- кількість електролитів у поті зменшується після 48-годинного використання глюкокортикоїдів за рахунок посилення реабсорбції у протоках нормальних потових залоз. При МВ рівень електролитів залишається підвищеним. Ця техніка може використовуватися у постпубертатному періоді, в якому частота хибнопозитивних результатів потового тесту вища.
Дослідження ДНК з ідентифікацією двох мутацій ТРБМ гену є абсолютним підтвердженням діагнозу муковісцидозу, а також має певне прогностичне значення. Слід пам’ятати, що невиявлення мутацій не означає відсутність захворювання.
Диференційна діагностика МВ:
Питання диференційної діагностики МВ є досить складним у зв’язку зі значним клінічним поліморфізмом цього захворювання. Необхідно диференціювати легеневий синдром МВ і хронічний бронхіт, а також такі спадкові захворювання, які супроводжуються ураженням легенів, як первинний імунодефіцитний стан, синдром Картагенера, Хаммена-Річа, дефіцит ААТ, ідіопатичний гемосідероз та інші. Синдром мальабсорбції при МВ слід диференціювати від целіакії, дисахаридазної недостатності, синдрому Швахмана, ферментопатій. Цироз печінки при МВ диференціють з цирозом печінки іншої етіології, який частіше всього розвивається після інфекційного гепатиту. У всіх випадках підтвердженням діагнозу МВ є позитивний результат потового тесту.
Лікування муковісцидозу:
Лікування хворого на МВ починається відразу після встановлення діагнозу і проводиться упродовж усього життя. Воно включає базисні заходи, що спрямовані на корекцію дисфункції органів і пов’язаних з цим симптомів, та заходи по лікуванню ускладнень. Позитивних результатів вдається досягти, у першу чергу, за рахунок чіткої організації виконання лікувально-реабілітаційної програми.
Головні принципи терапії хворих на МВ:
- зберігання функції легенів шляхом забезпечення евакуації бронхіального секрету та боротьби з інфекцією дихальних шляхів;
- нормалізація нутритивного статусу з корекцією панкреатичної недостатності, дієтичною компенсацією енерговитрат та недостатнього засвоювання компонентів їжі;
- підтримка функцій гепатобіліарної системи;
- психоемоційна адаптація;
Очищення бронхіального дерева від в’язкого мокротиння досягається через вплив фізичних та фармакологічних факторів. Фізичні методи евакуації мокротиння – перебування у спеціальних дренажних позиціях тіла з вібро-, кінетомасажем або без нього, вправи дихальної гімнастики, фізичні вправи, спорт. Ці процедури обов’язково проводити щоденно двічі, тричі на добу за індивідуальними програмами для кожного хворого, що розроблюються залежно від віку, важкості стану пацієнту, особливостей бронхолегеневого процесу.
У дітей грудного та молодшого віку використовуються пасивні методи евакуації мокротиння: статичні або динамічні дренажні позиції відповідно анатомії бронхіального дерева, позиційний дренаж у стандартних положеннях з періодичним масажем зони, що дренується. Така техніка може використовуватись і у дітей старшого віку, але більш ефективним є сполучення її з методами дихальної гімнастики, коли повітря використовується як поршень, що виштовхує згустки секрету із периферичних бронхів. Ми рекомендуємо починати заняття дихальною гімнастикою у вигляді гри вже з 2-3 річного віку, у повному обсязі з 6-7 років.
Значно покращує мобілізацію та видалення мокротиння “активний цикл дихання” з форсованим видихом. Він складається з 3 компонентів:
- контроля дихання – спокійне діафрагмальне дихання у звичайному обсязі з релаксацією верхніх відділів грудної клітки;
- вправи на збільшену екскурсію грудної клітки – максимально глибокі вдихи із спокійним видихом, повторюються 3-4 рази, у деяких пацієнтів одночасно з похлопуванням грудної клітки під час видиху;
- 1-2 форсованих видиха з участю м’язів черевної стінки зі звуком “х” (“хаффінг”) та повернення до контроля дихання.
Може проводитись сидячи або у дренажних позиціях. Повторюється до повного відділення мокротиння, але не більш 20-30 хвилин.
Ефективною вправою дихальної гімнастики є аутогенне тренування, що також проводиться циклами з 3 компонентів. Використання на протязі циклу різних за обсягом вдихів є ефективним способом евакуації секрета з різних відділів бронхів. Після вдиху проводиться спокійний видих, вправа повторюється 3-4 рази підряд з поступовим нарощенням дихального обсягу, потім слідує другий компонент – коротка пауза – відпочинок у вигляді спокійного дихання. Третьою складовою циклу є посилений, дмухаючий видих з відкашлюванням мокротиння.
Додаткове значення мають вправи з позитивним тиском під час видиху за допомогою спеціальної маски, флаттера. Вони протипоказані при загостреннях, значній емфіземі, кровохарканні, після легеневих кровотеч.
Видаленню мокротиння сприяють і постійні фізичні вправи, які при добрій толерантності повинні виконуватися щоденно. Це тренує м’язи, покращує емоційний фон. У першу чергу практичними навичками проведення процедур повинні оволодіти батьки хворих.
Фізіотерапія може модифікуватися, а під час загострення бронхолегеневого процесу та у випадках поганого відділення мокротиння повинна сполучатися з використанням муколітичних препаратів. Серед них найбільш ефективним залишається М-ацетилцистеїн у вигляді різних комерційних препаратів (ацетилцистеін – Росія, мукосольвін – Німеччина, флуімучил – Італія та ін.). Ацетилцистеїн швидко розріджує мокротиння завдяки вільній сульфгідрильній групі, яка розриває дисульфідні зв’язки мукопротеїдів. За останніми даними, його застосування може призвести до порушення функцій війок епітелію бронхів, це обмежує термін його використання двома – трьома тижнями. Найбільш ефективним є введення ацетилцистеїну інгаляційним шляхом: інгаляції 2-3 р. на добу по 15 хв. з застосуванням 1-4 мл 20% розчину. Для дітей від 0 до 3 років, коли інгаляційне введення ускладнено, можливо сполучати оральний та в/м’язовий шляхи введення. Використовується 10% розчин із розрахунку 10 мг/кг від 1 до 3 разів на добу. Хворим у вкрай важкому стані можна вводити препарат в/венно у вигляді 5% розчину (разова доза дітям до З років 50-100 мг, старше 3 років – 150-400 мг).
Ефективним є застосування препаратів амброксолу, якому властива здатність активізувати сурфактант альвеол, розріджувати бронхіальний секрет, стимулювати функцію циліарного епітелію бронхіол. Призначається у дозі 10-60 мг на добу відповідно віку. Шляхи введення: інгаляційний, оральний у таблетках та розчині, в/венний.
Досягнути значного муколітичного ефекту можна за допомогою інгаляцій теплого ізотонічного або гіпертонічного розчину (3-4%) хлористого натрію, 2-4% розчину гідрокарбонату натрію, лужних мінеральних вод за допомогою ультразвукових інгаляторів 2-3 рази на добу. Слід памятати, що прийом муколітиків передує фізіотерапіі.
У даний час у зарубіжних клініках для поліпшення мукоцілеарного кліренсу досить широко використовується рекомбінантна людська ДНКаза (Рulmozym). Дослідженнями доведено, що застосування ДНКази інгаляційно покращує функцію легень, зменшує частоту загострень.
Санаційна бронхоскопія може використовуватися тільки за умовою неефективності інших методів видалення бронхіального секрету. Показаннями до неї є: виражений обтураційний синдром, нерозправлені ателектази. У хворих з легеневим серцем вона протипоказана.
За наявності гіперреактивності бронхів доцільним є використання бронходилататорів (В2-агонистів, антихолінергічних препаратів, еуфіліну) у дозах, які застосовуються при лікуванні бронхіальної астми.
У період загострень лікування повинно здійснюватись в умовах спеціалізованого пульмонологічного відділення, слід уникати контактів з хворими на гостру респіраторну патологію. У зв’язку з перехресним інфікуванням рекомендується окрема госпіталізація хворих на МВ, колонізованих та неколонізованих синьогнійною паличкою.
Для боротьби з синьогнійною та іншою бактеріальною інфекцією, яка викликає прогресування легеневого процесу, застосовується антибактеріальна терапія. Основними принципами антибіотикотерапії при МВ є:
- використання препаратів згідно з чутливістю отриманої мікрофлори;
- мікробіологічний контроль терапії з кількісним визначенням збудників;
- агресивність терапії з призначенням комбінацій 2-3 антибіотиків у максимальному віковому дозуванні, пролонговано (до 1-1,5 міс.);
- основним шляхом введення препаратів у період загострення слід вважати внутривенний;
- профілактичне використання антибіотиків.
Таблиця 1.
Основні антибіотики, які використовуються для лікування МВ
| Амоксіцилін | |||
| Аугментін | |||
| Флуклоксациллін | |||
| Азлоцилін | |||
| Піперацилін | |||
| Тікарцилін | |||
| Карбеніцилін | |||
| Цефтазидим (фортум) | |||
| Цефоперазон (цефобид) | |||
| Цефтриаксон | |||
| Азтреонам | |||
| Іміпенем (тієнам) | |||
| Меропенем | |||
| Гентаміцин сульфат | |||
| Амікацин сульфат | |||
| Тобраміцин | |||
| Нетілміцин | |||
| Офлоксацин | |||
| Ципрофлоксацин | |||
Антибіотики широкого спектру дії призначаються при перших симптомах загострення з наступною корекцією за результатами мікробіологічного дослідження мокротиння. Безсимптомне одержання збудників у планових посівах мокротиння, які повинні проводитись один раз у 2-3 місяці, також є показанням до призначення відповідної антибактеріальної терапії.
Для ерадикації стафілокока застосовують оксацилін, флуклоксацилін, за неефективності їх замінюють або комбінують з макролидами, клиндаміцином, цефалоспоринами, котримоксазолом, ріфампіцином. При одержанні Н. influenzae призначують амоксіцилін, цефалоспорини тощо. Препаратами вибору при лікуванні синьогнійної інфекції залишаються аміноглікозіди у різних комбінаціях з β-лактамними антибіотиками (у першу чергу – цефалоспоринами III і IV генерації, новими формами пеніцилінів, у другу – з тієнамами та азтреонамом), похідними фторхінолонів. Дози багатьох антибіотиків підвищуються. Необхідно при ініціальній інфекції Ps. aeruginosa досягати стійкої ерадикації збудника. Така тактика запобігає формуванню резистентності флори та колонізації бронхів синьогнійною паличкою, яка є головним чинником розвитку хронічного запального процесу, значно погіршує життєвий прогноз.
З профілактичною метою ми рекомендуємо хворим з хронічною колонізацією Ps. aeruginosa регулярно інгаляційно аміноглікозиди або коліцин 2 рази на день у дозах у 2-3 рази перевищуючих добову до в/в введення протягом 7-10 днів один раз у два, три місяці, а також пероральне чи парентеральне призначення антибіотиків під час респіраторних вірусних інфекцій усім хворим на МВ.
При приєднанні алергічного бронхопульмонального аспергільозу призначається гормональна терапія у вигляді стероїдних гормонів – преднізолону з розрахунку 1-1,5 мг/кг маси на добу (дається у 2 прийоми – у 8 та 12 годин) протягом від 2-х тижнів до місяця, а у деяких випадках – довше. За наявності протипоказань (дистрофія, гнійно-септичні ускладнення і інші) проводиться лікування інгаляційними стероїдними препаратами (бекламетазон та інші). Протигрибкові препарати, як правило, ефекту не дають.
Дієта хворих на МВ відрізняється високою енергетичною цінністю, яка перевищує вікову потребу на 20-50%. Вона максимальна у хворих з низькими масо-ростовими показниками, а також під час загострень бронхолегеневого процесу. Збільшення енергоспоживання досягається за рахунок 2-3 додаткових прийомів висококалорійної їжі на протязі дня. По можливості, необхідно рекомендувати вільний вибір їжі, особливо дітям старшого віку і дорослим. Раціон хворих повинен якомога менше відрізнятися від того, який споживають здорові члени сім’ї.
У добовому раціоні жири забезпечують 35-45% калоражу, білки – 15%, вуглеводи – 45-50%. Недоцільно при достатній ферментотерашї обмежувати хворих у жирах, це викликає дефіцит есенціальних жирних кислот та жиророзчинних вітамінів. З сучасних позицій загальна кількість жирів має бути підвищеною, перевага надається рослинним жирам.
У хворих першого року життя слід орієнтуватися на грудне вигодовування, оскільки жіноче молоко містить ліпазу та есенціальні жирні кислоти. В інших випадках при застосуванні адаптованих сумішей слід забезпечити калорійність раціону не менше як 120-150 ккал/кг на добу, а у разі недостатнього збільшення маси підвищити до 150-200 ккал/кг. Доцільним є використання сумішей на напівелементній основі, які містять ліпіди з жирними кислотами скороченої довжини, так звані середньоланцюгові тригліцеріди – СЛТ (Прегестиміл, Пепті-Юніор, Нутраміген). У випадках первинної або вторинної лактазної недостатності показані низько- та безлактозні суміші.
Враховуючи те, що хворі втрачають багато хлорідів і натрію, кількість солі у дієті слід збільшити на 2-5 г на добу, особливо при підвищенні температури навколишнього середовища.
В основі корекції порушень процесів травлення лежить замісна терапія ферментами підшлункової залози. Лише застосування сучасних удосконалених форм ферментних препаратів – Креон (Solvay), Панцитрат (Knoll) дозволяє здійснити ефективну корекцію екзокринної недостатності, значно покращити нутритивний стан хворих завдяки можливості реалізації висококалорійної, високоліпідноі дієти.
Таблиця 2.
Склад мікросферичних ферментних препаратів (капсула)
| Креон (8000) | |||
| Креон (25000) | |||
| Панцитрат (10000) |
Ці ферментні препарати являють собою мікросферичні гранули з ентеророзчинною кислотостійкою оболонкою, які вміщені у желатинову капсулу. Перевагами цих препаратів на відміну від тих, що застосовувались раніше, є кислотостійкість, рівномірність розподілу ферментів в обсязі переварюваної їжі, 100% біодоступність, майже відсутність ускладнень. Вони легко дозуються дітям раннього віку без капсули, у цих випадках їх змішують з їжею, бажано з фруктовим пюре. Ферменти необхідно давати безпосередньо перед прийомом їжі.
Дозу ферментів слід визначати в спеціалізованому лікувальному закладі. Вона добирається індивідуально, залежно від ступеня синдрому мальабсорбції. Початкова доза складає 4000-6000 МО за активністю ліпази на кг ваги на добу. Таке дозування може бути достатнім, але частіше, у хворих з важким перебігом МВ, вираженою недостатністю функції підшлункової залози добову дозу поступово треба збільшувати до 8000-10000 МО на кг ваги під контролем клінічних симптомів та лабораторних ознак панкреатичної недостатності (поліфекалії, наявності стеатореї, тощо). Ферменти розподіляють між усіма прийомами їжі (основними і додатковими), разові дози розраховують пропорційно загальному вмісту жирів у раціоні.
Необхідність подальшого збільшення добової дози ферментних препаратів вище 10000 МО/кг потребує ретельного обстеження для визначення причин рефрактерності стеатореї та інших симптомів. Відомо, що дози більш 10000-15000 МО/кг підвищують ризик ускладнення – розвитку фіброзуючої колонопатії.
Як доповнюючий чинник при лікуванні мальабсорбції на фоні замісної терапії ферментами у підлітків з ознаками підвищеної секреції шлункового соку та рефрактерністю стеатореї використовуються антисекреторні препарати – блокатори Н2-рецепторів гістаміну.
Критеріями правильно підібраної дієти і дози ферментів є нормалізація кількості та характеру випорожнень, відсутність нейтрального жиру у тричі зробленій копрограмі, нормалізація показників екскреції фекального жиру до 5 г на добу, нарощення маси тіла. Дослідження авторів свідчать, що нормалізація трофіки дітей, хворих на МВ зменшує важкість перебігу захворювання, сприяє покращенню функції органів дихання.
Якщо не вдається нормалізувати нутритивний статус дитини цими засобами, а також при стійкому порушенні апетиту, виражених симптомах мальабсорбції додатково уживають ентеральне зондове харчування. Воно використовується у дітей різного віку, звичайно вночі, забезпечує 1/3 – 1/4 добового калоражу, вводяться стандартні дитячі або спеціальні суміші. Ферменти приймають перед зондовим харчуванням, у разі використання напівелементних сумішей з СЛТ разова доза зменшується наполовину. У тяжких випадках застосовують часткове або повне парентеральне харчування.
Для хворих на МВ необхідна корекція жиророзчинних вітамінів А та Е. Необхідні добові дози для ентерального призначення такі: віт. А – від 1000 до 5000 Од. в залежності від віку; віт. Е – новонародженим – 10 мг на добу; дітям до 3-х років – 30-50 мг; старшим 3 років – 50-100 мг, підліткам, дорослим – 200 мг.
Для корекції вмісту мікроелементів пропонуються наступні склади у добових дозах: цинк – 10-15 мг, молібден – 250-300 мг, мідь – 2-3 мг, марганець – 3-4 мг, хром – 100-150 мкг, селен – 100-125 мкг, йод – 100-150 мкг. З цією метою пропонується використовувати препарати, які вміщують полівітамінні комплекси і мікроелементи у необхідному дозуванні (Мультітабс, Юнікап М, Юнікап Т, Дуовіт, Мультіфіт, Нутрісан, Супрадін та інш.), багато з яких вміщують також бета-каротин, який є антиоксидантом.
Враховуючи порушення процесів травлення, проведення дуже частої масивної антибактеріальної терапії, що призводить до відхилень у складі кишкової мікрофлори, доцільним є використання у комплексній терапії біопрепаратів з цілющою мікрофлорою – біфідо-, лактобактеріями (Біфіформ, Лінекс та інші), а також продуктів харчування, збагачених ними (кефір, йогурт).
При розвитку синдрому дистальної інтестинальної обструкції призначається ацетілцистеін перорально 3-4 рази на день та у пряму кишку (100 мл 10% розчину) на протязі 3-4 днів. Рекомендується збільшений прийом рідини, корекція дієти, оцінка адекватності ферментотерапії.
З метою профілактики та лікування уражень гепато-біліарної системи показано призначення урсодеоксіхолевої кислоти (препарати Урсофальк, Урсосан), якій властива холеретична дія. Добова доза становить 15-20 мг/кг, розподілених на 3 прийома під час їжі. З профілактичною метою рекомендується вживати її 2-3 рази на рік протягом місяця дітям старшим трьох років, а при наявності ознак ураження печінки – майже постійно, роблячи перерви двічі на рік по місяцю. Додатково для покращення жовчоутворюючої та жовчовидільної функцій печінки можуть застосовуватись такі препарати, як алохол, холензім, фламін, холосас – курсами по 1-1,5 міс. Рекомендується застосування препаратів, що покращують метаболізм у гепатоцитах, стабілізують клітинні мембрани: есенціале, карсил по 1,5-2 міс. З-4 рази на рік.
Нагляд за хворими на МВ:
Лікування та нагляд за хворими МВ проводиться упродовж усього життя. Мається на увазі організація етапного лікування, яка включає Центри з діагностики і лікування МВ, місцеві лікувальні заклади, санаторії.
Центр по МВ здійснює діагностику захворювання, реєстрацію та спеціальний облік хворих, лікування у період загострення, регулярні обстеження для оцінки стану пацієнтів, їх фізичного розвитку, призначає терапію, яка буде проводитись в умовах районної поліклініки, дому, санаторію, проводить навчання батьків. До Центру дитина повинна поступати не менше 2-х разів на рік планово, а також при загостреннях.
У районних дитячих поліклініках повинно проводитись регулярне лікарське спостереження за хворими та сестринський патронаж. Частота профілактичних лікарських оглядів:
- на перших трьох місяцях життя – 4 рази на місяць;
- на першому році життя – 2 рази на місяць;
- у наступні роки – щомісячно при важкому перебігу МВ; 1 раз у 2 місяці при середньо-важкому і сприятливому перебігу МВ.
Дільничний лікар і сестра організовують реабілітаційні дії відповідно рекомендаціям Центру, проводять наступні лікувально-організаційні заходи:
- систематичне спостереження за виконанням призначеної лікувально-реабілітаційної програми;
- діагностику загострень та ускладнень захворювання;
- профілактичні щеплення;
- лікування інтеркурентних захворювань;
- лабораторні дослідження (мікробіологічне дослідження мокротиння, інші – при необхідності);
- профілактику і лікування анемії, гіповітамінозів;
- огляд спеціалістами: стоматологом – 2 рази за рік; ЛОР-лікарем – 2 рази за рік; іншими спеціалістами – при необхідності.
Після виписки із стаціонару необхідно щоденно виконувати дренажні вправи, масаж, дихальну гімнастику, лікувальну фізкультуру за призначеною програмою, продовжувати постійний прийом ферментних препаратів, підібраних у відповідній дозі, додержуватись дієти.
Рекомендується практично постійний прийом полівітамінних препаратів (бажано з мінеральними додатками) курсами по 1-1,5 міс. з 2-х тижневою перервою, зі зміною препаратів; призначаються вітаміни А та Е у вищевказаних дозах по 1 міс. З-4 рази на рік, вітаміни К і Р – тільки за необхідністю.
У період ремісії при важкому варіанті МВ з наявністю хронічної синьогнійної інфекції в легенях ми рекомендуємо профілактичні курси антибіотиків у вигляді інгаляції, доцільно використання аміноглікозидів і поліміксину В (коліцину) у двократному добовому дозуванні. Інгаляцію бажано проводити після фізичних методів евакуації мокротиння. При респіраторних вірусних інфекціях, які звичайно сприяють загостренням, також призначають антибіотики.
Дуже важливим питанням в житті хворих МВ є проблема імунізації. Літературні дані, а також тривалий досвід нашого спостереження за хворими свідчать на користь обов’язкової імунізації за існуючою загальновизнаною схемою, з урахуванням деяких особливостей: імунізація повинна проводитися тільки в період стійкої ремісії, не раніше 1 місяця після останнього загострення.
Направлення дітей, хворих на МВ, у дошкільні дитячі установи, як правило, не показано. Однак, багато з них відвідують школу, а тому лікар повинен бути чітко орієнтованим відносно захворювання, щоб вести адекватне спостереження за цими хворими з метою розробки оптимального режиму відвідування школи (надання додаткового вихідного дня в середині тижня, скорочення навчального дня). Діти з важкою формою захворювання підлягають навчанню удома.
Профілактика муковісцидозу, генетичне консультування та допологова діагностика:
Впровадження досягнень молекулярної генетики у практику генетичного консультування і допологової діагностики започаткувало нову еру профілактики муковісцидозу в родинах високого ризику народження дитини, хворої на муковісцидоз, значно розширило діагностичні можливості генетичного консультування на підставі ідентифікації первинного генетичного дефекту. Це дозволяє використовувати результати тестування на мутації у батьків, близьких родичів хворої дитини і партнерів гетерозиготних носіїв гена муковісцидозу при розрахунках генетичного ризику.
В умовах значної гіподіагностики МВ в Україні, важливим для його ефективної профілактики є своєчасне встановлення діагнозу і проведення генетичного консультування в родині з використанням результатів молекулярно-генетичних досліджень (ДНК аналізу) як батьків, так і дитині. Досвід свідчить, що така тактика сприяє підвищенню діагностичної інформативності родин, які консультуються, до 95,5-98% , що дасть змогу використовувати ДНК аналіз у допологовій діагностиці. Проте в цих випадках можливість запобігти народженню хворої дитини існує тільки при повторних вагітностях.
Молекулярно-генетичний аналіз повинен містити в собі як пряму детекцію гена, так і сімейний аналіз поліморфних ДНК маркерів, тісно зчеплених із геном МВ. На Україні ці дослідження виконуються в Інституті молекулярної біології та генетики НАН України, м. Київ.
З метою проведення проспективного генетичного консультування необхідно проводити молекулярно-генетичний скринінг мутацій, так званий “каскадний”, серед близьких родичів пробанда, в якого вдалося ідентифікувати мутації, а також у батьків хворої дитини і їхніх партнерів при повторних шлюбах. Якщо в результаті тестування виявлена відсутність мутації у другого партнера, це дозволяє модифікувати ризик гетерозиготного носійства від загальнопопуляційного 1:24 до 1:50 і ризик мати хвору дитину стає 1/50 х 1/4 = 1/200 (0,5%) при частоті мутації delF508 – 56%.
На сучасному етапі для пренатальної діагностики МВ використовується пряма детекція гена, а також сімейний аналіз зчеплених поліморфних ДНК маркерів із локусом гена трансмембранного регулятора білка (ТРБМ). У випадках, коли родина діагностично не інформативна, пренатальна діагностика проводиться методом визначення ферментів мікроворсинок кишечника плода в амніотичній рідині у строк вагітності 17-18 тижнів (Інститут акушерства і гінекології ім. Отта, м. Санкт-Петербург, лабораторія пренатальної діагностики спадкових хвороб – керівник проф. В.С. Баранов).
На даному етапі вирішення проблеми МВ у нашій країні допологова діагностика стала ефективним методом боротьби з цією хворобою. Її метою є уточнення наявності цього важкого спадкового захворювання у плода. Одержання матеріалу для допологової діагностики проводиться за допомогою інвазивних досліджень, таких як: біопсія хоріону або плаценти та амніоцентез.
У діагностично інформативних родинах допологову діагностику можливо проводити за допомогою біопсії хоріону у І триместрі вагітності, або за допомогою плацентоцентезу у II триместрі. У частково інформативних родинах допологову діагностику необхідно проводити у два етапи. В І триместрі у плода визначається наявність (або відсутність) мутантної хромосоми. При її відсутності МВ у плода виключається. При наявності мутантної хромосоми вагітність за бажанням жінки може бути перервана (вірогідність МВ 50%), або продовжена з обов’язковою біохімічною діагностикою у строк 17-18 тижнів.
Для генетичного консультування родин групи високого ризику МВ, а також проведення допологової діагностики у цих же родинах, вони можуть бути направлені у відповідні медичні заклади (інститут ПАГ АМН України). Найбільш ефективним є консультування родини та планування допологової діагностики до вагітності, що сприяє своєчасному та максимальному використанню усіх методів допологовоі діагностики.
Поряд з пренатальною діагностикою та генетичним консультуванням, раціональна профілактика МВ у масштабах міста, регіону, а можливо, у віддаленому майбутньому і всієї держави повинна включати відповідні скринінгові програми як серед новонароджених, так і інших вікових груп, спрямовані на активне виявлення хворих МВ (новонароджені), так і гетерозиготних носіїв (інші вікові групи).
В останній час все більше уваги привертає скринінг новонароджених на МВ з використанням молекулярно-генетичних методів. Цей підхід вже знайшов застосування в економічно розвинених країнах з високим рівнем мутації delF508 (Великобританія, країни Скандинавії, Данія) та визначених мажорних мутацій – не менше 80-85%. За інших умов він вважається нерентабельним.
Висновки:
Уявлення про МВ як про летальне захворювання у більшості випадків несправедливе. До цього висновку приходять усі лікарі і дослідники, які займались вивченням МВ достатньо тривалий час. Своєчасна, максимально рання діагностика, адекватна терапевтична тактика та безперервний нагляд за хворою на МВ дитиною запобігають виникненню багатьох симптомів та відстрочують розвиток ускладнень та незворотних змін в організмі. Впровадження останніх досягнень сучасної медицини, що викладені у методичних рекомендаціях, у практичну діяльність лікарів різних фахів, які працюють з хворими на МВ, підвищить ефективність надання медичної допомоги цій категорії хворих.
Використання панкреатичних ферментів у вигляді мікрогранул у ентеророзчинній оболонці за визначеними принципами робить набагато ефективнішою замісну корекцію екзокринної недостатності підшлункової залози, що поряд з відповідною дієтою сприяє значному покращенню нутритивного та загального стану хворих на МВ.
Сучасні методи, спрямовані на евакуацію бронхіального секрету та тактика наступальної антибактеріальної терапії зумовлюють більш активний вплив на розвиток хронічного інфікування дихальних шляхів високопатогенними збудниками, що зменшує частоту загострень, дозволяє максимально зберегти функцію дихальної системи, що обумовлює тривалість життя хворих на МВ.
Застосування холеретичних препаратів (урсодеоксихолева кислота) високо ефективне у профілактиці та лікуванні уражень гепатобіліарної системи при МВ.
Цілком вірогідно, що без створення чіткої системи диспансерного нагляду за хворими МВ ефективність одно-дворазового перебування в Центрах МВ буде зведено до нуля. Належним чином організована чітка система нагляду за хворими на МВ створює умови для адекватної терапевтичної тактики, генетичного консультування, пренатальної діагностики, санітарно-просвітньої роботи з батьками, навчання їх необхідним навичкам по догляду за дитиною.
Центр МВ діагностує захворювання, надає лікарську допомогу і складає план реабілітаційних заходів та підтримуючої терапії на подальший період. Роль поліклініки, районного пульмонолога, дільничного лікаря полягає в контролі за виконанням рекомендацій, запобіганні загострень, проведенні постійної підтримки хворої дитини і членів її сім’ї, які зазнають впливу негативних соціальних, психологічних і матеріальних факторів.
Література:
- Капранов Н.И., Рачинский С.В. Муковисцидоз. М., 1995.- 188 с.
- Михайлец Л.П. Муковисцидоз: генетическое консультирование и дородовая диагностика //Журнал практического врача.- № 2.- 1997.- C. 27-29.
- Молекулярно-генетичний аналіз муковісцидозу в Україні /Л.А. Лівшиць, С.А. Кравченко, М.В. Нечипоренко ті інші //Матеріали науково-практичної конференції “Сучасний стан медичної генетики в Україні”.- К., 19-20 травня 1999 р.- С. 99-100.
- Резник Б.Я., Бабий Й.Л., Лившиц Л. А. Муковисцидоз у детей й подростков. Одеса: Чорномор’я, 1994.- 141 с.
- Резник Б.Я., Старец Е.А. Современные представлення о муковисцидозе //Діагностика та лікування.- 1998.- № 1.- С. 24-27.
- Columbo С, Battezzati PМ, Podda М et al. Ursodeoxycholic acid for liver disease associated with cystic fibrosis: a double-blind multicenter trial. Hepatology 23;1996:1484-1490.
- Conway SP. Recombinant human DNase (rhDNase) in cystic fibrosis: it is cost effective? Archives of Disease in Childhood 77;1997:1-3.
- Cystic fibrosis – Current Topics. Vol. 3 /Edited by Dodge J, Brock D, Widdicomble J.- John Willey & Sons, New York – 1996:320.
- Genetic Testing for Cystic Fibrosis. NIH Consensus Statement Online. 1997, Apr. 14-16, № 106.
- Hodson М, Gedds D. Cystic fibrosis. Chapman – London. 1995:439.
- Lanng S, Hansen A, Thorstemsson B, Nerup J, Koch C. Glucose tolerance in patients with cystic fibrosis: five year prospective study. British Medical Journal 1995;311:655-659.
- Lee I, Ip W, Durie P. Is fibrosing colonoparthy an immune mediated disease? Archives of Disease in Childhood 1997;77:66-70.
- MacDonald A. Nutritional Management of cystic fibrosis. Archives of Disease in Childhood 1996;74:81-87.
- Practical Guidelines for Cystic Fibrosis Care /Edited by Catherine М.Нill. London, 1998:203.
- Ramsey В. Management of Pulmonary Disease in Patients with Cystic Fibrosis. The New England Journal of Medicine 1996;335(3):180-188.
- Wallis C. Diagnosing cystic fibrosis: blood, sweat and tears. Archives of Disease in Childhood 1997;76:85-91.
- Zielenski J, Tsui LC. Cystic fibrosis. Genotypic and Phenotypic Variation. Annual Review of Genetic 1995;29:777-807.
Переглянуто редакційною колегією I.B.I.S.: 15/02/2002
Дивіться також:
Розміщення вуха відносно очей (продовження)
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”


На верхньому графіку – 3, 50 та 97 перцентилі значення розміщення вуха відповідно до ваги при народженні. Нижній графік показує 3, 50 та 97 перцентилі значення у відсотках розміщення вуха відповідно до ваги при народженні. Вимірювання проводились у 87 доношених (48 хлопчиків та 39 дівчаток) та 111 недоношених (55 хлопчиків та 56 дівчаток) ізраїльських новонароджених гестаційного віку 27-41 тижнів. Вимірювання проводились у перші 36-60 годин життя. Жодна дитина не мала вроджених вад. Merlob P et al: Birth Defects: Orig Art Series XX(7):5, 1984. Sivan Y et al: J Med Genet 22:414, 1985.
Розміщення вуха відносно очей
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”
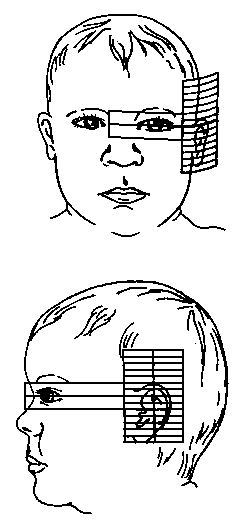
3, 50 та 97 перцентилі розміщення вуха відносно рівня очей відповідно до гестаційного віку. Визначення рівня вух щодо рівня очей – це вимірювання відстані між найвищою точкою вуха та рівнем внутрішнього кута ока. Рівень внутрішнього кута очей визначається горизонтальною лінією між двома внутрішніми кутами. Вимірювання проводиться з використанням прозорої плівки з нанесеною на неї міліметровою шкалою. Центральна горизонтальна лінія розміщується на рівні внутрішніх кутів очей. Щоб визначити відстань між рівнем внутрішнього кута і найвищою точкою вуха, міліметрова шкала повинна покривати вухо. Техніка виконання за Feingold M et al: Birth Defects: Orig Art Series 10(13):3, 1974. Див. пояснення нижче. Merlob P et al: Birth Defects: Orig Art Series XX(7):17, 1984.


3, 50 та 97 перцентилі процентного значення розміщення вуха відповідно до рівня очей відповідно до гестаційного віку.
Вухо над рівнем очей
Загальна довжина вуха
Вимірювання проводились у 87 доношених (48 хлопчиків та 39 дівчаток) та 111 недоношених (55 хлопчиків та 56 дівчаток) ізраїльських новонароджених 27-41 тижнів гестації у перші 36-60 годин життя. Жодна дитина не мала вроджених вад. Merlob P et al: Birth Defects: Orig Art Series XX(7):5, 1984. Діаграми вперше були надруковані Sivan Y et al: J Med Genet 20:213, 1983.

Вимірювання довжини вуха
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”

3, 50 та 97 перцентилі довжини вуха, що визначається, як найбільша відстань між крайніми верхньою та нижньою точками вушних раковин, відповідно до гестаційного віку. Вимірювання проводились у 87 доношених (48 хлопчиків та 39 дівчаток) та 111 недоношених (55 хлопчиків та 56 дівчаток) ізраїльських новонароджених 27-41 тижнів гестації у перші 36-60 годин життя. Жодна дитина не мала вроджених вад. Merlob et al: BirthDefects: Orig Art Series XX(7):5, 1984.


3, 50 та 97 перцентилі довжини вуха відповідно до ваги при народженні. Вимірювання проводились у 87 доношених (48 хлопчиків та 39 дівчаток) та 111 недоношених (55 хлопчиків та 56 дівчаток) ізраїльських новонароджених 27-41 тижнів гестації у перші 36-60 годин життя. Жодна дитина не мала вроджених вад. Sivan Y et al: J Med Genet 22:414, 1985.
Вимірювання відстані між зіницями
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”.
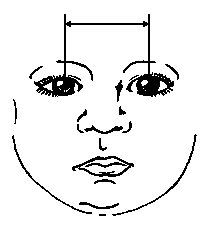
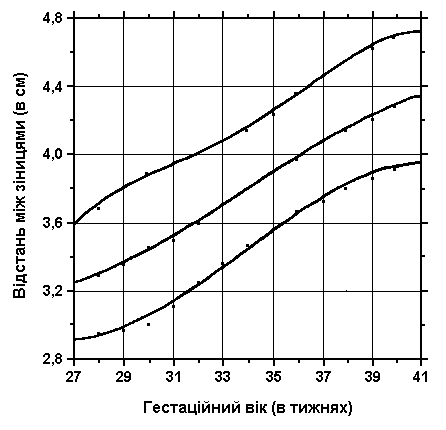
3, 50 та 97 перцентилі вимірів між зіницями очей відповідно до гестаційного віку. Дані обстеження 87 доношених (48 хлопчиків та 39 дівчаток) та 111 недоношених (55 хлопчиків та 56 дівчаток) новонароджених 27-41 тижнів гестації у перші 36-60 годин життя. Жодна дитина не мала вроджених вад. Міжзінична відстань потім була вирахувана за формулою: Відстань між зіницями = 0,7+0,59 * відстань між внутрішніми кутами очей + 0,41 * відстань між зовнішніми кутами очей. Merlob P et al: Birth Defects: Orig Art Series XX(7):11, 1984.
Довжина очних щілин (продовження)
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”

3, 50 та 97 перцентилі довжини очної щілини відповідно до гестаційного віку. Вимірювання проводились у 200 доношених (100 хлопчиків та 100 дівчаток) та 60 недоношених новонароджених європеоїдної раси 32-38 тижнів гестації у перші 2-3 дні життя дітей в спокійному стані. Jones KL et al: J Pediatr 92:787, 1978.

3, 50 та 97 перцентилі довжини очної щілини (відстань між двома кутами ока) відповідно до гестаційного віку. Вимірювання проводились у 87 доношених (48 хлопчиків та 39 дівчаток) та 111 недоношених (55 хлопчиків та 56 дівчаток) ізраїльських новонароджених 27-41 тижнів гестації у перші 36-60 годин життя. Жодна дитина не мала вроджених вад. Merlob et al: Birth Defects: Orig Art Series XX(7):5, 1984.
Довжина очних щілин
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”.
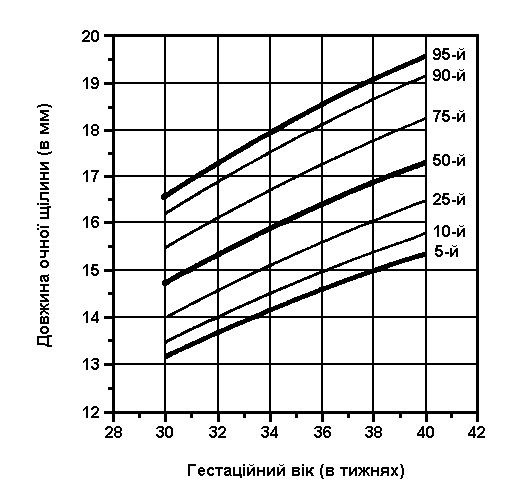
Середнє значення та перцентилі довжини очної щілини новонароджених 30-40 тижнів гестації за Thomas, Gaitantzis та Frias (J Pediatr 111:267, 1987). Антропометричні дані 200 доношених білих американських дітей та 60 недоношених дітей 2-3 днів життя за Jones, Hanson, Smith (J Pediatr 92:787, 1978).
Расові відмінності в довжині очних щілин при народженні
випадків |
значення (в см) |
значення ± 1 SD |
||
|---|---|---|---|---|
Порівняння значень та стандартних відхилень довжини очної щілини латиноамериканців, темношкірих та білих новонароджених в перші 3 дні життя в спокійному стані. Значення отримані при огляді 120 латиноамериканських та 101 темношкірого новонароджених дітей 37-42 тижнів гестації, народжених в Нью-Йорку. Не знайдено жодної різниці між правими і лівими очима та різними статями. За Fuch та співавтор. (J Pediatr 96:77, 1980). Порівняння білих дітей за Jones та співавтор. (J Pediatr 92;787, 1978).
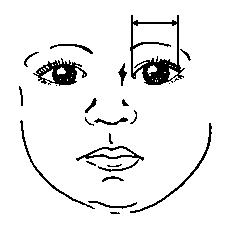
Вимірювання відстані між очима
Із українського перекладу книги “Growth References: Third Trimester to Adulthood. Compiled by Saul RA, Geer JS, Seaver LH, Phelan MC, Sweet KM, Mills CM. Greenwood Genetic Center. 1998”

3, 50 та 97 перцентилі відстані між внутрішніми кутами очних щілин нанесені на графік відповідно до гестаційного віку. Вимірювання проводились у 87 доношених (48 хлопчиків та 39 дівчаток) та 111 недоношених ізраїльських новонароджених з гестаційним віком від 27 до 41 тижнів. Вимірювання проводились у перші 36-60 годин життя. Жодна дитина не мала вроджених вад. Merlob P et al: Birth Defects: Orig Arts Series XX(7):5, 1984.
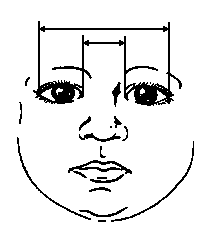

3, 50 та 97 перцентилі відстані між зовнішніми кутами очних щілин нанесені на графік відповідно до гестаційного віку. Див. пояснення вище. Merlob P et al: Birth Defects: Orig Arts Series XX(7):5, 1984.
|
|
|
|
|
|





