
Хвороба Гірке
(Von Gierke Disease)
Тетяна Дмитрівна Загорулько
Лікар-неонатолог
Волинського обласного дитячого територіального медичного об’єднання
Загальний огляд глікогенозів:
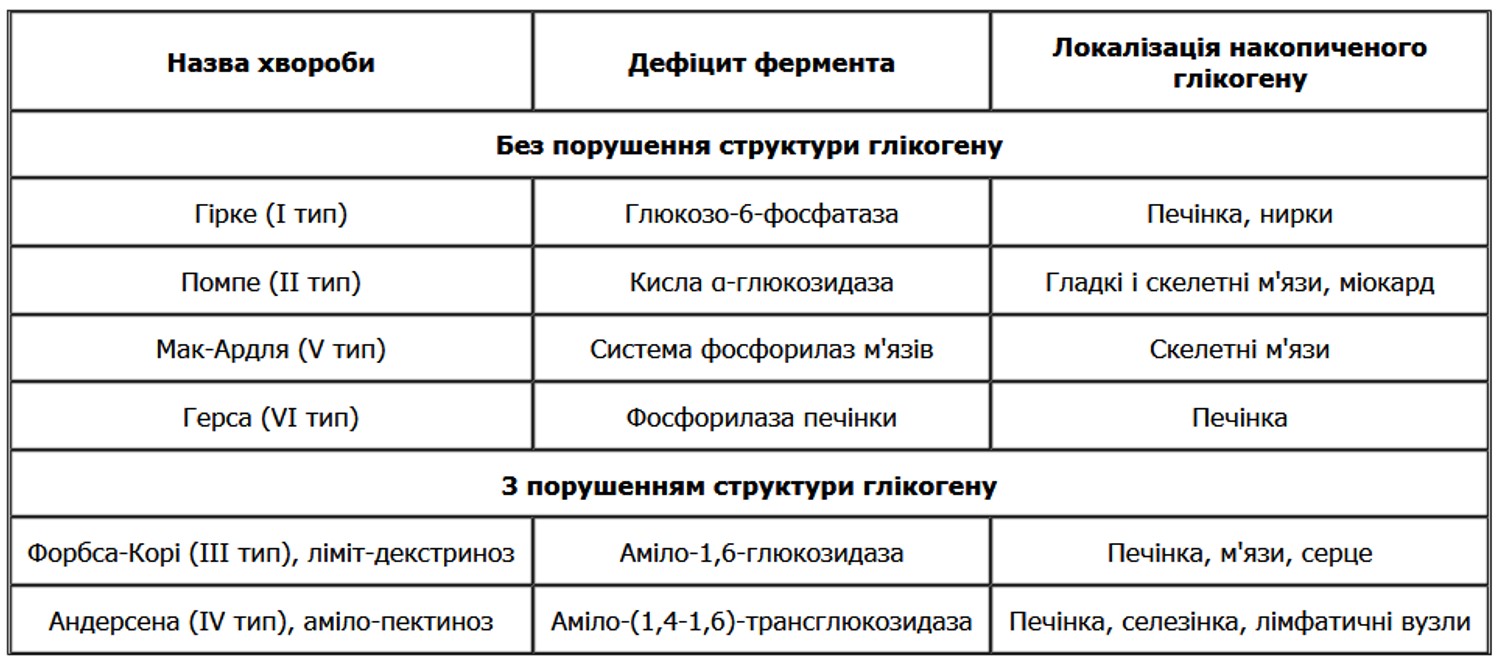
Основні запаси глікогену знаходяться в печінці і скелетних м’язах. Глікоген печінки і м’язів використовується в залежності від потреби організму (лабільний глікоген). Глікоген нервових клітин, провідної системи серця, ендотелію, епітеліальних покривів, слизової оболонки матки, сполучної тканини, ембріональних тканин, хрящів, лейкоцитів є необхідним компонентом клітин, і його вміст не піддається помітним коливанням (стабільний глікоген). Регуляція обміну вуглеводнів здійснюється нейроендокринним шляхом: гіпоталамус, гіпофіз (АКТГ, тиреотропний, соматотропний гормони), β-клітини підшлункової залози (інсулін), наднирники (глюкокортикоїди, адреналін) і щитовидної залози. Порушення вмісту глікогену проявляються в зменшенні або збільшенні кількості його в тканинах і появі там, де він, як правило, не виявляється. Ці порушення найбільш виражені при цукровому діабеті і при спадкових вуглеводних дистрофіях – глікогенозах. Останні зумовлені відсутністю або недостатністю фермента, що приймає участь в розщепленні депонованого глікогену, і тому належать до спадкових ферментопатій, або хвороб накопичення. На даний час добре вивчені 6 типів глікогенозів, зумовлених спадковою недостатністю шести різноманітних ферментів. Це хвороби Гірке (І тип), Помпе (II тип), Мак-Ардля (V тип) і Герса (VI тип), при яких структура накопиченого в тканинах глікогену не порушена, і хвороби Форбса-Корі (III тип), і Андерсена (IV тип), при яких вона різко змінена.
Глікогенози
Визначення:
Хвороба Гірке (синоніми: недостатність глюкозо-6-фосфатази, глікогеноз типу І, глікогенна гепатонефромегалія) – спадкова ферментопатія, зумовлена відсутністю фермента глюкозо-6-фосфатази, що приймає участь в розщепленні депонованого глікогену, наслідком цього дефіциту є накопичення глікогену в печінці і нирках. Синдром вперше був описаний Гірке у 1929 році, як поєднання збільшеної печінки і порушення метаболізму глікогену. Захворювання зумовлене дефектами генетичного матеріалу.
Діагностичні критерії:
Відставання в зрості, гепатомегалія, гіпоглікемія, гіперлактатацидемія.
Частота:
Глікогеноз І типу зустрічається з частотою 1 випадок на 100 тис. новонароджених. Питома вага хвороби Гірке серед усіх глікогенозів становить 25 відсотків. Однак, клінічний перебіг саме глікогенозу І типу вважається найважчим.
Тип успадкування:
Аутосомно-рецесивний.
Етіологія:
Глікогеноз І типу спричинений дефектом глюкозо-6-фосфатази, яка каталізує розпад глюкозо-6-фосфату на фосфат і глюкозу. Ця реакція відіграє ключову роль в регуляції глікемії, оскільки вона контролює кінцевий крок в процесах глікогенолізу і глюкогенезу. В нормі глюкозо-6-фосфат, який утворюється в процесі метаболізму глікогену, в печінці під дією глюкозо-6-фосфатази перетворюється у вільну глюкозу, яка надходить у кров і використовується в інших органах; в м’язах, які не містять глюкозо-6-фосфатази, глюкозо-6-фосфат використовується для власних енергетичних потреб, окислюючись аеробним або анаеробним шляхом до лактату.
В 1952 році Корі довів, що хвороба спричинена дефіцитом глюкозо-6-фосфатази. Пізніше дослідження показали, що у деяких пацієнтів не спостерігається дефіциту глюкозо-6-фосфатази, хоча багаточисельні функціональні тести демонструють неспроможність гідролізувати глюкозо-6-фосфат in vivo. Цей стан був названий глікогенозом типу І. Припускається, що він зумовлений дефіцитом глюкозо-6-фосфатспецифічної транслокази, яке забезпечує вхід глюкозо-6-фосфатази (G6Pase) в пластинки ендоплазматичного ретикулуму.
Отже, недостатність глюкозо-6-фосфатази (G6Pase) спричинює глікогеноз типу Іa, a дефіцит глюкозо-6-фосфаттранслокази (G6PT1) – глікогеноз типу Іb. Першу гіпотезу про механізм виникнення глікогенозу типу Іb сформулював Arion. Nordlie описав пацієнта з глікогенозом типу Іс, з дефіцитом транслокази, специфічної до фосфату (G6PT2), яка сприяє проникненню фосфату за межі ендоплазматичного ретикулуму в цитоплазму, завершуючи таким чином гідроліз глюкозо-6-фосфату.
Існування глікогенозу типу Іd, властивого людям з дефіцитом транслокази (G6PT3), здатної виводити глюкозу з ендоплазматичного ретикулуму, ніколи не було доведене достовірно. Ген, відповідальний за G6Pase, локалізований на хромосомі 17q21; за глюкозо-6-фосфат-транслоказу – на 11 хромосомі (11q23).
Патофізіологія:
Печінка не здатна виконувати свою функцію як глюкозо-гомеостатичного органу. Швидке падіння рівня глюкози в крові, що в нормі спричиняється в перші декілька годин після народження (рефлекторне споживання материнської глюкози), не може компенсуватись звичайними гомеостатичними механізмами, таким чином дефіцит глюкози зростає. Наслідком тяжкої гіпоглікемії (іноді глюкоза взагалі не визначається в крові) можуть бути судоми, апное, ціаноз, ушкодження ЦНС. Дефіцит глюкозо-6-фосфатази зумовлює утворення з глюкозо-6-фосфату лактату. Лактатемія прямо пропорційна ступеню стимуляції розпаду глікогену. Акумуляція молочної кислоти в крові спричиняє ацидоз з великою аніонною різницею.
Anion gap = [Na+] – ([Сl] + [НСО3]) = 12 ± 4 мекв/л
В нормі концентрація аніонів в крові дорівнює концентрації катіонів, зменшення в плазмі бікарбонатів збалансовано підвищенням концентрації інших аніонів. Збільшення величини “anion gap” свідчить про збільшення в крові невимірюваних аніонів, тобто про ацидоз. Величезне збільшення фосфорильованих проміжних компонентів гліколізу конкурентно пригнічує рефосфориляцію аденінових нуклеотидів, сприяє деградації нуклеїнових кислот, наслідком чого є збільшення в крові сечової кислоти. Гіперурикемія може досягати рівня, що вимагає застосування інгібіторів ксантиноксидази для попередження нефролітіазу. Тяжка гіпоглікемія стимулює секрецію гормонів мозкової речовини наднирників, які активують ліпопротеїнліпазу і звільняють кількість жирних кислот. Останні в свою чергу транспортуються до печінки, де використовуються для синтезу тригліцеридів і виводяться з печінки як ліпопротеїди низької густини. Навіть при значній гіпоглікемії у пацієнтів з І типом глікогенозу не розвивається значний кетоз, оскільки надлишок ацетил-коензиму А (СоА), який виникає внаслідок гліколізу, активує ацетил СоАкарбоксилазу, що стимулює утворення малоніл СоА на першому етапі синтезу жирних кислот. Малоніл пригнічує транспорт жирних кислот в мітохондріях, тому їх β-окислення і виділення енергії для підтримки клітин не виникає. Ці причини поглиблюють зменшення рівня глюкози і пояснюють відсутність кетонових тіл.
Припускають, що причиною носових кровотеч є пошкодження синтезу мембранних глікопротеїдів, хоча це не дає вичерпного пояснення щодо дефекту агрегації тромбоцитів.
При глікогенозу типу Іb пацієнти є сприйнятливі до грам-позитивної флори. Для нейтрофілів у хворих глікогенозом типу Іb характерний послаблений кисневий вибух на інфекційний стимул. Це зумовлює нейтропенію та зниження індивідуальної стійкості до інфекцій. При кисневому вибусі утворюється супероксид – головний засіб захисту від грам-позитивної флори.
Клініка:
Діти відразу після народження виглядають здоровими, та хвороба маніфестує вже з перших днів життя у вигляді гепатомегалії. Печінка протягом першого року життя м’яка при пальпації, потім стає щільною, вузлуватою. Зовнішній вигляд хворого характерний: маленький зріст – постійна ознака хвороби Гірке, повні щічки, кругле лялькоподібне обличчя, ксантоми (зумовлені підшкірним відкладанням жиру), виступаючий живіт, що різко контрастує з тонкими кінцівками. На шкірі можна побачити численні екхімози, пацієнт скаржиться на часті носові кровотечі, що є наслідком дисфункції тромбоцитів. М’язи атрофовані. Можуть бути присутні ознаки подагри (з боку суглобів, нирок). Хворі відстають у статевому дозріванні, хоча фертильність не порушується, у жінок, хворих глікогенозом І типу, описані успішні вагітності, які були завершені кесарським розтином на 35 тижні гестаційного розвитку і народженням здорових дітей. Хворі не адаптовані до голодування, внаслідок гіпоглікемії можуть виникнути судоми. Внаслідок відкладення глікогену в нирках, останні також збільшуються в розмірах.
Виникнення ускладнень можна затримати за допомогою контролю за метаболічними процесами. Серед ускладнень виділяють: печінкова аденома з ризиком малігнізації, анемія, остеопенія (переломи), кисти яйників.
Маніфестація ниркових ускладнень розпочинається з прихованої клубочкової гіперфільтрації, яка передує розвитку протеїнурії, що може перерости в ниркову недостатність. Поширені гіперкальційурія, нефрокальциноз і камені в сечовивідних шляхах.
Пошкоджена функція нирок у деяких хворих є причиною виникнення артеріальної гіпертензії.
Вважається, що дисліпідемія може стати причиною виникнення панкреатиту, атеросклерозу та серцево-судинних ускладнень. Легенева гіпертензія – рідкісне ускладнення і прогноз його виникнення надзвичайно несприятливий.
Глікогеноз типу Іb має подібну клінічну картину. Крім ознак, характерних для типу Іа, типу Іb властиві нейтропенія, порушення функції моноцитів, що сприяє розвитку частих вторинних інфекційних захворювань, виразковуванню слизових кишківника та ротової порожнини, розвитку запальних кишкових захворювань.
Діагностика:
- Загальний аналіз крові: анемія, нейтропенія.
- Першочергові лабораторні дослідження мають включати визначення глюкози в крові, електроліти.
- Дослідження функцій печінки і нирок; кількість сечової кислоти в крові, креатиніну, кліренс сечовини, креатиніну, рівень білірубіну та трансаміназ.
- Коагулограма – зміни характерні для порушення функцій тромбоцитів (кількість тромбоцитів збільшена), зменшення фібриногену та інших факторів зсідання крові.
- УЗД нирок, печінки.
- Морфологічна діагностика того чи іншого типу можлива при біопсії з допомогою гістоферментативних методів.
- Інші методи:
- проба з глюкагоном (1 мг/м² поверхні тіла): гіпоглікемія не зменшується, проте збільшується кількість лактату.
- проба з галактозою (1,75 г/кг) не спричиняє змін глюкози в крові, хоча зростає кількість молочної кислоти.
Лікування:
В лікуванні важливе місце посідає дієта, скерована на уникнення метаболічних розладів, запобігання ускладненням з боку нирок і печінки, досягнення зросту згідно вікової норми. Лікування складається з частого годування, безперервного нічного ентерального живлення, вживання вуглеводів, що повільно всмоктуються і обмеження вживання фруктози і галактози, які можуть посилювати гіперлактатемію. Калорійність добового раціону має бути суворо контрольована. Недостатнє харчування не відкорегує метаболічні порушення (гіпоглікемію, гіперлактатемію, гіперурикемію); надлишкове харчування збільшує накопичення глікогену, гепатомегалію, гіперліпідемію, спричинює ожиріння. Дієта має містити велику кількість важкозасвоюваних вуглеводів (60-65% ккал від добової калорійності) і малу кількість ліпідів (20-25% від добової калорійності).
Для новонароджених і малят корисні часті годування і введення безперервно через назогастральний зонд їжі, забезпечуючи спочатку 8-10 мг глюкози/кг/хв, потім 5-7 мг/кг/хв.
Додаткова терапія включає вітаміни, кальцій для попередження остеопенії і аллопуринол, коли присутня гіперурикемія.
Для попередження ускладнень з боку нирок ефективним є застосування інгібіторів ксантинонидази. Для корегування анемії застосовують препарати заліза.
Для вагітних цей препарат безпечний.
Корегувати нейтропенію при глікогенозі тип Іb може гранулоцитарно-макрофагальний колонієстимулюючий фактор, що зменшує тяжкість бактеріальних інфекцій і полегшує перебіг запальних кишкових явищ.
Filgrastim (G-CSF, Neupogen) активує, стимулює продукцію, дозрівання, міграцію і цитотоксичність нейтрофілів. Доза 5 мкг/кг для дітей та дорослих. Протипокази: гіперчутливість до препарату. Взаємодія: синергічний ефект з ІЛ-3 (збільшення кількості мегакаріоцитів і тромбоцитів). Вагітність: безпечність для вагітних не доведена. Застереження: не застосовувати 12-24 год. після цитотоксичної хіміотерапії, оскільки підвищується чутливість мітотичних мієлоїдних клітин до хіміотерапії; не вживати з содою; застосовувати обережно при подагрі, псоріазі. Побічні ефекти: гарячка, біль в кістках, грипоподібні симптоми.
Ефективність лікування оцінюється за допомогою моніторування таких показників – крива росту, ступінь гепатомегалії, артеріальний тиск, біохімічних параметрів – рівень глюкози в крові більше 3,5 ммоль/л, лактат сечі менше 0,6 ммоль/л в нічній і денній сечі, тригліцеридемія, холестеринемія, урикемія, протеїнурія і загальний аналіз крові.
Лікування ускладнень полягає у трансплантації печінки після безуспішної терапії дієтою. Трансплантація печінки корегує гіпоглікемію та інші біохімічні показники, але не нормалізує кількість нейтрофілів, також не доведено, що це може попередити ураження нирок. Трансплантація нирок, проведена у випадках тяжкої ниркової недостатності, не корегує гіпоглікемії. Можливість одночасної пересадки печінки і нирок обговорюється, але такі операції не проводились. Під час хірургічних втручань, з огляду на ризик виникнення кровотеч і метаболічного дизбалансу необхідний постійний моніторинг глікемії, лактацидемії, системи згортання крові. Глікемія має підтримуватись перфузіями 10% глюкози перед, протягом і під час оперативного втручання. Заборонено застосовувати розчини, що містять лактат (наприклад розчин Рінгера). Пацієнти, які пройшли терапію, і у яких було досягнуте клінічне і лабораторне видужання, підлягають диспансерному нагляду і повинні проходити повний медичний огляд кожні 6 місяців.
Акушерська тактика:
Небезпека виникнення кровотеч і гіпоглікемії потребують постійного моніторування вагітностей для запобігання мимовільним викидням і мертвонародженням. Рекомендовано розродження шляхом кесарського розтину приблизно на 35 тижні вагітності.
Диференційний діагноз:
Інші типи глікогенозів, гепатомегалія іншої етіології.
Прогноз:
Залежить від дотримання дієти, запобігання виникненню ускладнень.
Номер з каталогу МІМ:
232200 Glycogen Storage Disease I.
Література:
- Deeksha Bali, Yuan-Tsong Chen, Stephanie Austin, Jennifer Goldstein. Glycogen Storage Disease Type I (https://www.ncbi.nlm.nih.gov/books/NBK1312/).
- Jones KL. Smith’s Recognizable Patterns of Human Malformation. Philadelphia: W.B.Saunders Company 1997:680.
Переглянуто редакційною колегією I.B.I.S.: 16/08/2004
|
|
|
|
|
|





